A Draft Human Pangenome Reference
Nature May 9 2023
10.1038/s41586-023-05896-x
The human pangenome – one of the most complete collections of genome sequences released so far – captures rich human diversity

Researchers have released a high-quality collection of reference human genome sequences – the human pangenome – that better captures diversity from different human populations compared to the current human reference genome.
The work was led by the international Human Pangenome Reference Consortium (HPRC), a group funded by the National Human Genome Research Institute (NHGRI), part of the National Institutes of Health (NIH) and consisting of 14 institutes, including EMBL’s European Bioinformatics Institute (EMBL-EBI).
Genome sequences differ only slightly among individuals. In the case of humans, any two genomes are, on average, more than 99% identical. Small genomic differences contribute to each person’s uniqueness and can provide insights about their health, helping to diagnose disease and guide medical treatments.
These small differences also mean that using one standard reference genome, as many studies currently do, can have limitations. Expanding the range of genomes to increase the diversity present in the human reference genome will help progress personalised medicine by enabling clinicians to better tailor treatment to individual patients.
This draft human pangenome reference includes the maternal and paternal genome sequences from 47 people, and the researchers are aiming to increase this number to 350 by mid-2024. The work, published in the journal Nature, is one of several papers published today by HPRC members. The majority of the genomes used to create the human pangenome reference were collected as part of the 1000 genomes project, the largest public catalogue of human variation and genotype data from a wide range of populations.
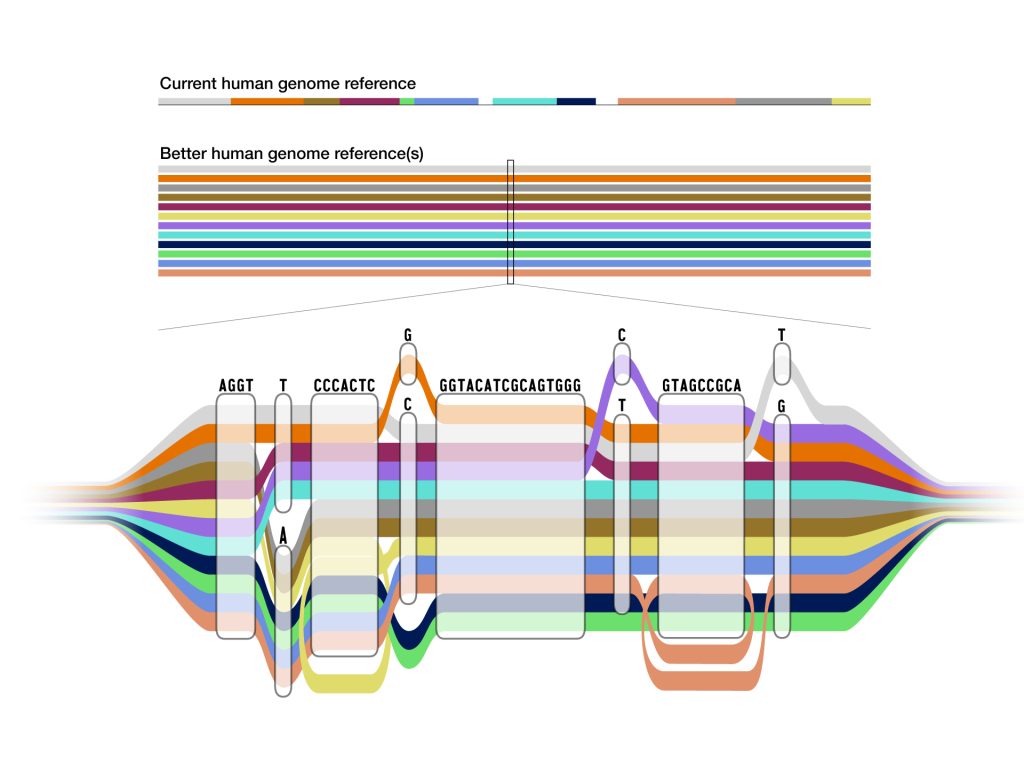
To understand genomic differences between humans, scientists create reference genomes – a digital amalgamation of human genome sequences that can be used as a comparison base to align, assemble, and study other human genomes.
The original reference human genome is nearly 20 years old and has been regularly updated as technology advances and researchers fix errors and discover more genomic regions. However, it is fundamentally limited in its representation of human diversity as it consists of genomes from around 20 people, and most of the reference sequence is from only one person.
“Everyone has a unique genome, so using a single reference genome sequence for every person can lead to inequities in genomic analyses,” said Adam Phillippy, Senior Investigator in the Computational and Statistical Genomics Branch within NHGRI’s Intramural Research Program, and a co-author of the main study. “For example, predicting a genetic disease might not work as well for someone whose genome is quite different from the reference genome.”
While the previous reference genome sequence was single and linear, the pangenome represents many different versions of the human genome sequence at the same time. This gives researchers a wider range of options for using the pangenome in analysing other human genome sequences.
“By using the pangenome reference, we can more accurately identify larger genomic variants called structural variants,” said Mobin Asri, PhD student at the University of California Santa Cruz and co-first author of the paper. “We are able to find variants that were not identified using previous methods that depend on linear reference sequences.”
Structural variants – regions of DNA that include genomic inversions, insertions, or deletions – can involve thousands of bases. Until now, researchers have been unable to identify the majority of structural variants that exist in each human genome using short-read sequencing due to biases inherent in using a single reference sequence. The advent of new long-read sequencing technologies has been transformative for projects such as this, allowing researchers to capture the information within a genome in a more accurate way.
“The use of long-read sequencing will close gaps and resolve highly repetitive and difficult-to-characterise regions,” said Heather Lawson, Assistant Professor in the Department of Genetics, Washington University in St Louis. “This, combined with the collective genomes of hundreds of diverse individuals, will broaden the catalogue of variation to study in the context of human health and disease. This will inform clinical variant interpretation and also open the door for functional studies of variants falling in the complex regions that have remained largely uncharacterised in genomic studies.”
In order to understand the differences in the genes present across the individual genomes represented in the human pangenome, researchers in EMBL-EBI’s Ensembl team needed to map the high-quality annotations on the reference human genome generated as part of the GENCODE project, across the pangenome. This was challenging, as each genome has a slightly different set of genes, with some individuals having more or fewer copies of certain genes. In some cases, a gene that is functional in the reference genome might be broken in an individual genome or vice-versa.
“There’s no question that the reference human genome has revolutionised our understanding of human biology and health,” said Fergal Martin, Eukaryotic Annotation Team Leader at EMBL-EBI. “Despite this, the current reference is an artificial genome, with many gaps and errors, and is a biased representation of our genetic diversity as a species. Our reference pangenome represents a stepping stone to addressing these issues in a structured and scalable manner. Over time this will allow for far more precise representations of genomes from populations across the globe and ultimately, improved diagnosis of disease, more targeted approaches to treatment, and better clinical outcomes.”
The human pangenome data are openly accessible on the Ensembl human pangenome project page and through Ensembl Rapid Release.
The human pangenome reference is a work in progress; researchers from the international HPRC plan to continually add more genome sequences to increasingly improve the quality of the pangenome reference.
This news item was adapted from the NHGRI press release.
Nature May 9 2023
10.1038/s41586-023-05896-x