Towards an expression atlas for an entire brain
Researchers who study how genes are expressed across a given tissue can now examine thousands of genes at once at cellular resolution, thanks to new methods developed at EMBL and published in Nature Biotechnology. The new techniques can be applied to a broad range of organisms, and expand the resources available for evolution-and-development research.
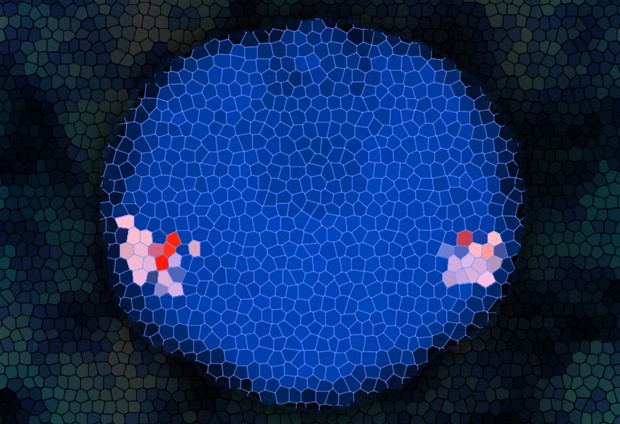
John Marioni of EMBL-EBI and Detlev Arendt of EMBL Heidelberg set out to augment an atlas of gene expression in the brain of a marine worm, Platynereis dumerilii, using single-cell RNA sequencing (RNAseq). The original atlas, constructed by the Arendt group, represents the expression patterns of around 170 genes in the worm brain at a specific point in development, all linked to their location.
Organisms and tissues are not just a random mash of different types of cells – they are organised in a manner that facilitates specific functions. To understand how they work, it is important to establish both their genetic profiles and their precise spatial organisation.
Marioni explains: “Single-cell sequencing generates expression profiles on a large scale, but these profiles are dissociated from the cells’ spatial location. That has been incredibly useful for discovering new cell types, but to understand how gene expression varies within cell types in living things, we do need to link each cell’s full expression profile with its specific location. That will allow us to explore, for a particular gene, how its expression profile changes across a tissue – and that will tell us a lot about how cell types function, how they interact with one another and how they evolved.”
To understand how gene expression varies within cell types in living things, we do need to link each cell’s full expression profile with its specific location.
In 2010, Detlev’s group published a meticulous 3D map of gene expression patterns in a relatively small number of genes. To augment the resource with thousands of genes expression profiles, the team back-mapped single-cell genomics data to the original atlas.
“In our in situ hybridisation experiments, we knew exactly where the cells were coming from. We were able to determine their expression patterns computationally, but on a fairly modest scale,” says Arendt. “To grasp what is going on throughout the tissue, we needed to be working on a much larger scale, and single-cell RNAseq offered that possibility. But with typical single-cell RNAseq experiments, you don’t have that detailed knowledge of where the cells are coming from.”
“We had data on the expression patterns of thousands of genes, but to find out where those cells are located spatially we had to compare their profiles to those in the Atlas for every cell we ran,” adds Marioni. “That told us which location in the reference atlas each cell most closely matched.”
The method allowed the researchers to match quantitative and spatial data, increasing by several orders of magnitude the number of markers that indicate specific cell types within a tissue. As a result, a wider pattern can start to emerge.
“My group focuses on this particular species to understand how the brain has evolved, but you could apply our method to any system that has a good reference gene expression database,” says Arendt.
You could apply our method to any system that has a good reference gene expression database.
“From the technical point of view, the simplicity and robustness of the method is very appealing – we achieved very high precision with a relatively low number of reference genes,” says Kaia Achim, a fellow in EMBL’s Interdisciplinary Postdocs (EIPOD) programme. “Potentially, our method could be used to investigate how and in which aspects gene expression patterns are stereotypical across different individuals and even species.”
“This will make a big difference in the resources available to smaller research communities that study evolution at a very fundamental level,” says Marioni. “The technology will help people gain much deeper insights into what’s going on in these non-model systems, and tell us a lot about how animals have evolved and what is mutable in nature. And that is pretty exciting.”
This post was originally published on EMBL-EBI News.