Pol III: Completing the family album
“The family gallery is now complete!” says Christoph Müller with a grin, having finally succeeded in resolving the 3D atomic structure of the largest and most elusive RNA polymerase, Pol III. After years of using X-ray crystallography as his structural biology method of choice, cryo-electron microscopy delivered the final breakthrough, thereby filling a glaring hole in the polymerase family album.
RNA polymerases are enzymes responsible for producing stretches of RNA, central to the working of the cell. All higher organisms, from yeast to humans, have three RNA polymerases, each made up of at least twelve subunits. Whilst the first 3D structures were solved around the start of the millennia, the largest family member, RNA polymerase III, or Pol III, has kept its secrets for itself – until now.
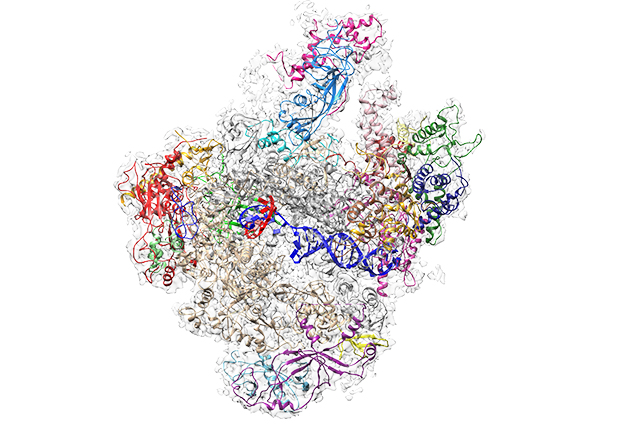
In a paper published today in Nature, Christoph Müller’s and Carsten Sachse’s groups present the first high-resolution structure of Pol III, achieved with cryo-electron microscopy (cryo-EM) techniques. “When we produced the first Pol III structure using cryo-EM in 2007, the resolution was so low it did not give us much detailed information – although we could locate the subunits that are specific to Pol III” explains Müller. A second structure with slightly better resolution followed in 2010, but it was the new generation of electron microscopy techniques and instrumentation that enabled Müller and his co-authors to improve the resolution so much that they now have a detailed picture of the enzyme.
in the last few years we’ve seen a real ‘resolution revolution’
For many years X-ray crystallography has been the structural biologist’s main tool to study the atomic structure of proteins and other biological molecules. This method involves firing intense X-ray beams at crystals of molecules. While this is a very powerful tool, it has its limitations – in particular the requirement for crystals. Convincing proteins to form crystals is no simple feat, and indeed some proteins – particularly large and complex proteins – cannot be convinced at all. Cryo-EM, which uses an accelerated beam of electrons to illuminate a sample, has the advantage that molecules can be studied in a more natural environment. “Cryo-EM is capable of revealing the atomic detail of biomolecules, and in the last few years we’ve seen a real ‘resolution revolution’,” says Sachse. “In the past, the technique was sometimes discounted as ‘blobology’, because in most cases you could only get an outline of the molecule. But the combination of improved imaging by direct electron detectors and better image processing algorithms have brought about a great leap in terms of the details achievable. I think this project really shows what cryo-EM can do, particularly for large protein complexes” he adds.
Trained as a crystallographer, Müller initially used crystallography to try to unlock the secrets of Pol III; but while his group and others have solved Pol I and II structures this way, success with crystallising Pol III remained elusive. “We just never managed to get crystals,” he says. “Looking now at the complete structure we can see that some parts are quite flexible, which may explain why it’s so hard to get these molecules to assemble correctly into a regular lattice, to make a crystal”.
Arjen Jakobi, a postdoc in the Sachse group, contributed to the project with computational analysis of the EM data and the building of an atomic model. “At the time we started these experiments there were no good tools available that would allow you to build a high-quality model making use of the full extent of the experimental data from electron microscopy” explains Jakobi. “So we developed protocols that build on the well-established methods of model refinement from X-ray crystallography, but that are tailored towards the specific challenges and experimental differences we have in electron microscopy.” “As a crystallographer, it is really surprising how much detail the cryo-EM structures reveal,” says Müller. “We can now build very precise models, with detailed information right down to the individual atoms.” The scientists make this point in today’s paper, too, comparing the EM structures with structures from crystallography of a similar resolution. “It is clear from these structures that cryo-EM is on par with X-ray crystallography which has long been considered the gold standard in structure determination.” adds Sachse. “And I think it is fair to say that more technological developments are under way to make cryo-EM even more powerful in the future.”

With the family of three now complete, Müller and his group can begin to make comparisons, understanding what makes each polymerase unique and what similarities they share. “We were able to resolve the structure of Pol III in two different states,” explains Müller group PhD student Niklas Hoffmann, who performed the EM experiments, “one with, and one without DNA in place, so we can really see in detail how the DNA is fed into and held by Pol III for transcription.” In order to produce the stretches of RNA, the polymerase uses one strand of the DNA helix as a blueprint for building a strand of RNA – this process is called transcription. For each letter of DNA, the enzyme pieces the complementary RNA letters together – A -U, T-A, G-C and C-G. Surprisingly, while the whole DNA is held quite tightly in place, the DNA-RNA duplex in the active centre has quite a bit of space to move. This is in contrast to Pol II where the DNA sits tighter in the active centre. “This possibly reflects that Pol III has to quickly produce lots of short RNA pieces, so it needs to release DNA quickly and doesn’t take time to bind it very tightly”, explains Hoffmann. The scientists were also surprised at how simple the termination of the transcription process is in Pol III when compared to Pol I and II. “In Pol III you only need a simple stretch of 5 ‘T’s in the genetic code to tell the enzyme to stop transcribing” says Hoffmann.
Oh, there is a lot more to be getting on with!
Comparisons also reveal why and how the enzymes differ in size. The smallest, Pol II, has 12 subunits and so recruits additional helper molecules to do the tasks it can’t; some of these associated molecules are already integrated in the Pol I and III complexes. “The advantage with having these parts built in is that the enzyme can start work straight away and doesn’t depend upon there being enough helper molecules around to lend a hand,” explains Müller. “As each molecule lines up to help the polymerase, it will be checked to see if it’s the right molecule, appearing in the correct order, etc. The disadvantage of having a complete ready-to-go complex may be that the processes are not so regulated,” he adds. “Being so key to cell growth and proliferation, Pol I and Pol III have to be carefully controlled, and if this goes wrong, it can be associated with cancer”.
So is there anything left to do now that the family gallery is complete? “Oh, there is a lot more to be getting on with!” says Müller. “We are interested in understanding how the whole process is initiated, how exactly it is terminated, how the process is regulated… these structures open up many new questions!”