New method offers broader and faster detection of protein-ligand interactions
EMBL scientists improve a protein analysis technique, significantly expanding its use and making it 100 times faster – a development that could accelerate drug discovery and fundamental biological research
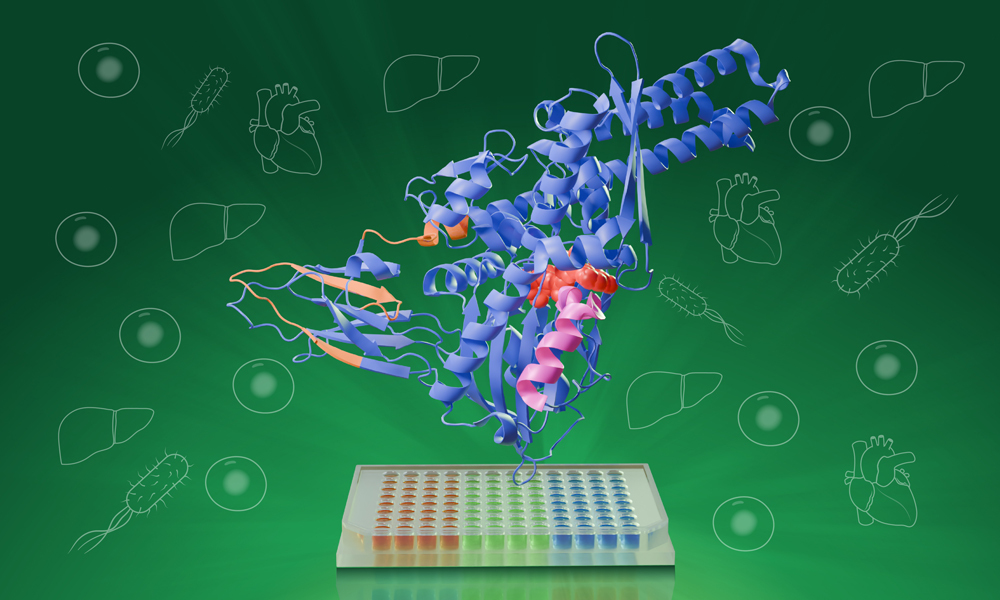
HT-PELSA identifies protein-ligand interactions by tracking how ligand binding affects protein local stability. Along with shifting the process to a 96-well format, this new tool can also work directly with crude cell, tissue, and bacterial lysates. Credit: Daniela Velasco/EMBL
Summary
HT-PELSA (high-throughput peptide-centric local stability assay), a new tool developed by EMBL researchers, speeds up sample processing 100-fold, making it much more effective at finding ligand-binding regions than previous methods.
Unlike the earlier version, HT-PELSA works directly with various biological samples, including crude cell, tissue, and bacterial lysates, allowing the detection of previously inaccessible protein targets, like membrane proteins – which account for the majority of all known drug targets.
By making large-scale protein research more affordable and efficient, HT-PELSA could accelerate drug discovery and transform how scientists study protein function in living systems.
Swedish chemist Jöns Jacob Berzelius, in a letter to a fellow chemist, first suggested the name ‘proteins’ for a particular class of biological substances, deriving it from the Greek word proteios, meaning ‘primary’ or ‘of first importance.’ Although scientists in the 1830s knew very little about proteins, it was already clear how essential they were to living organisms.
Long-known as the ‘workhorses of the cell,’ proteins are responsible for powering nearly every function in the body. Often critical to this is their interactions with other small molecules known as ligands. In a new study published inNature Structural and Molecular Biology, EMBL researchers introduce HT-PELSA, a high-throughput adaptation of an earlier tool that detects these interactions. This new tool can process samples at an unprecedented scale, a breakthrough that promises to accelerate drug discovery and our understanding of fundamental biological processes.
Still a fairly new tool itself, the original PELSA (peptide-centric local stability assay) method, launched last year by researchers at the Dalian Institute of Chemical Physics, Chinese Academy of Sciences, in collaboration with the Shanghai Institute of Materia Medica, identifies protein-ligand interactions by tracking how ligand binding affects protein stability. When a ligand binds to a protein, that part of the protein becomes more stable and less prone to the effects of enzymes like trypsin, which cuts proteins into smaller peptide fragments.
What made PELSA especially noteworthy was its ability to detect peptide-level changes in stability across the entire proteome – that is, across all of the proteins in an organism. Although effective, nearly every step in the PELSA workflow is done by hand, meaning scientists can only process a few samples at a time. This not only requires a lot of time and effort but also increases the risk of contamination and accidental error.
HT-PELSA streamlines this process significantly by shifting from full-size tubes to micro-wells. Such a change enables automation of PELSA’s steps and allows researchers to analyse hundreds of samples in parallel while maintaining the same sensitivity and reproducibility.
“Before, I could only do at most, maybe 30 samples per day,” said Kejia Li, first author of the study and postdoctoral fellow in the Savitski Team at EMBL. “Now, with HT-PELSA, we can scan 400 samples per day – it has highly simplified the workflow”. Before joining EMBL, Li was one of the developers of the original PELSA method.
While in PELSA, trypsin-cleaved peptides are separated from whole proteins based on their mass, HT-PELSA leverages the water-repellant nature of proteins. It utilises a surface that proteins stick to more readily than peptides, thus allowing the scientists to separate the two. This not only further automates the process, but also enables the detection of membrane proteins that, up until now, were hard or even impossible to study.
Membrane proteins, which make up around 60% of all known drug targets, are often difficult to extract without altering their structure or function. By working directly with complex samples, HT-PELSA can reveal how these proteins interact with potential drugs in their natural environment.
“It gives us a much more complete view of the proteome-ligand interaction landscape,” said Isabelle Becher, co-author of the study and Laboratory Officer in Charge in the Savitski Group. “You can see how these interactions are changing and get a real sense of the underlying biology”. By understanding protein-ligand interactions more deeply, scientists can create drugs that bind selectively to their target proteins, making treatments more effective and safer.
“HT-PELSA really opens the door for high-throughput understanding of protein function as well as accelerating drug development,” said Mikhail Savitski, Team Leader at EMBL Heidelberg and senior author of the study. “This is critical for understanding basic biology, discovering disease mechanisms, and for developing safer, more effective medicines”.
In the study, the team also demonstrates that HT-PELSA can detect changes in protein-protein interactions from ligand binding. In future studies, they hope to expand this to detect protein-protein and protein-nucleic acid interactions as well, which would further accelerate our understanding of the molecular organisation of the cell.