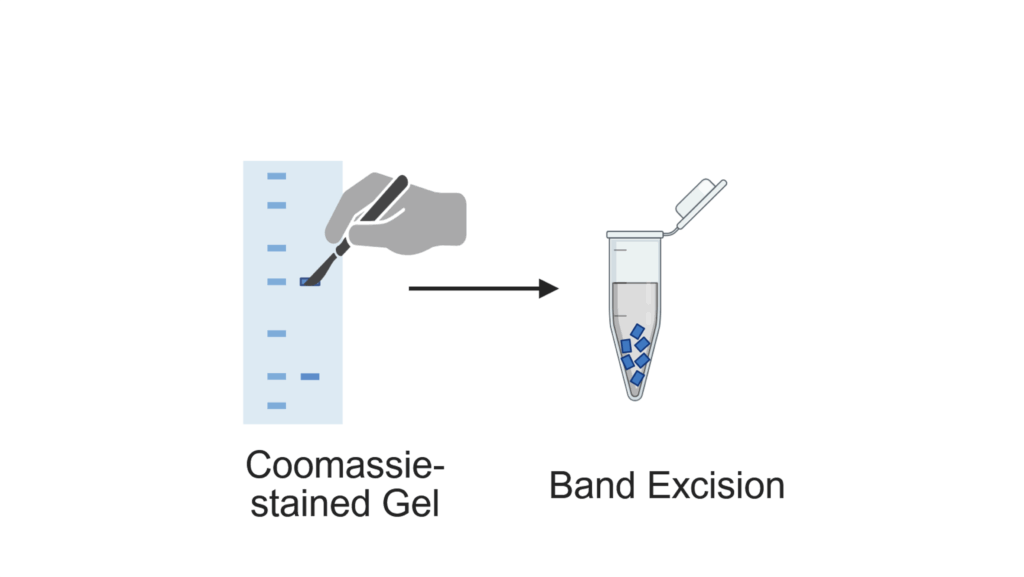
Protein identification
In-gel or in-solution
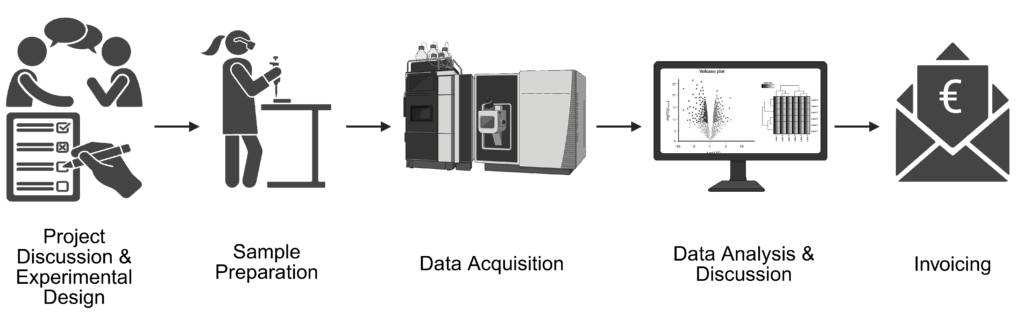
Empowering research through mass spectrometry-based proteomics
We provide a full service for your mass spectrometry-based proteomics experiment. This includes expert advice on your experimental design, sample preparation as well as processing and measuring of samples and data analysis including statistical analysis and data visualization.
For information on our services, browse our state-of-the-art analyses below. If you’re unsure whether we can support your experiment, please reach out — we’ll be able to provide you with more information.
Please also see our FAQs for more information.
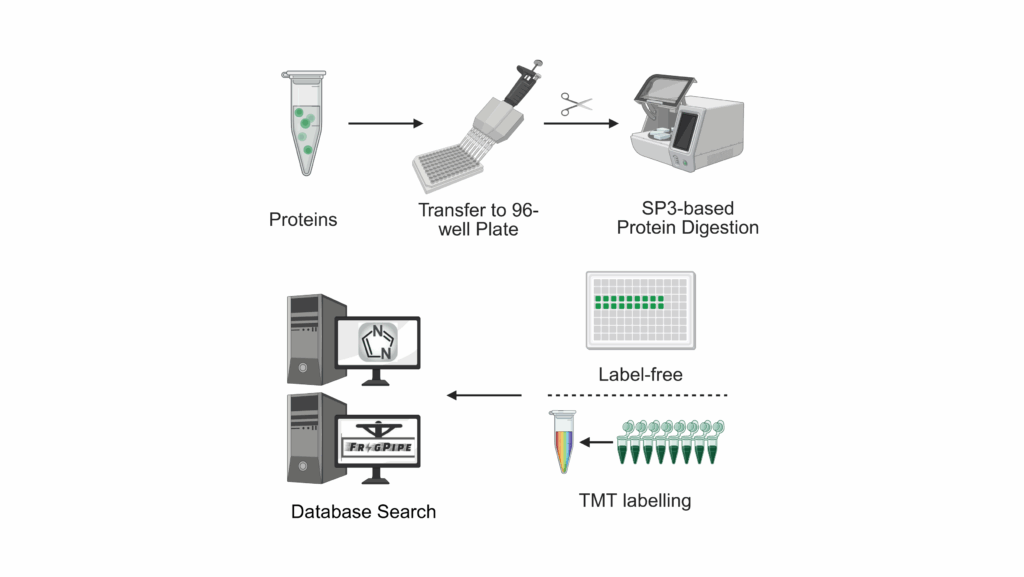
In-gel or in-solution
TMT- or DIA- based
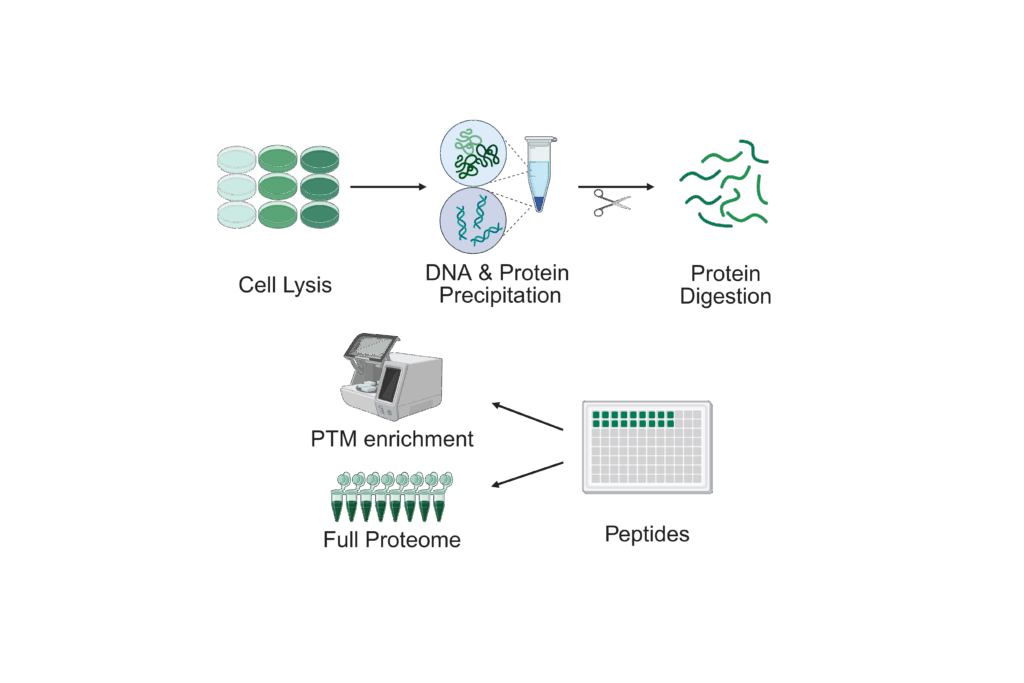
From purified proteins or on global scale
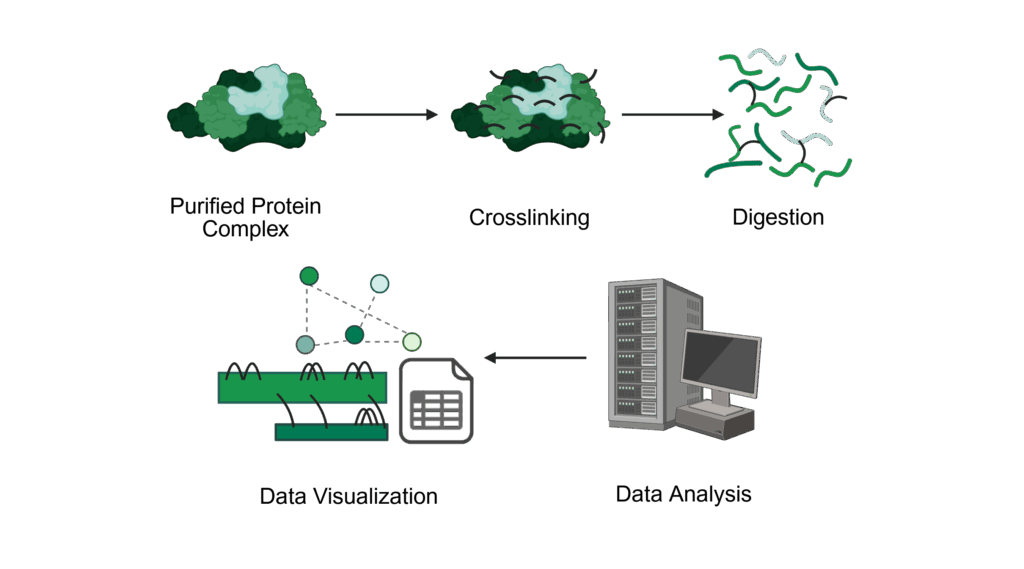
Protein interactions in purified complexes
| Consumables | EURO € |
|---|---|
| Sample processing fee (per sample) | 3 |
| TMT6-plex labelling reagent | 100 |
| TMT10/11-plex labelling reagent | 200 |
| TMT16/18-plex labelling reagent | 280 |
| Phosphoenrichment (per sample) | 20 |
| Desalting (per sample) | 20 |
| Offline high pH reverse phase chromatography (per sample) | 20 |
| Peptide-size exclusion chromatography (per sample) | 20 |
| Measurement time (per hour) | EURO € |
|---|---|
| Academia | 100 |
| Industry | 150 |
In order to estimate the total price for a specific type of a proteomics experiment such as, for instance, a full proteome analysis, please contact the PCF (pcf@embl.de) to discuss.
We provide protein identification, verification of mutations, inserts, and PTM analysis of purified proteins and protein complexes in-gel or in-solution samples.
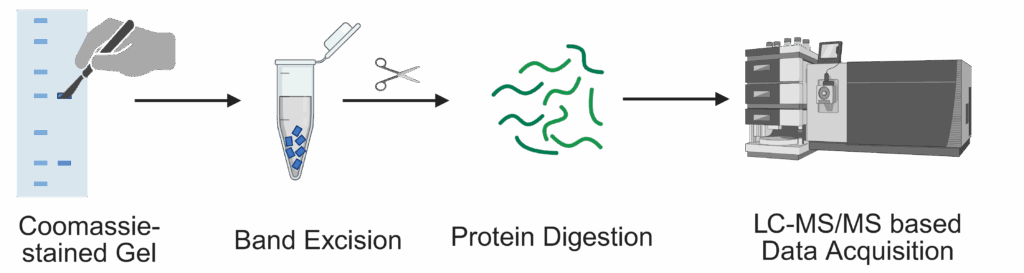
We provide in-gel based analysis of proteins for:
For this, we offer proteolysis by:

We also provide in-solution processing of samples for identification of proteins and protein complexes.
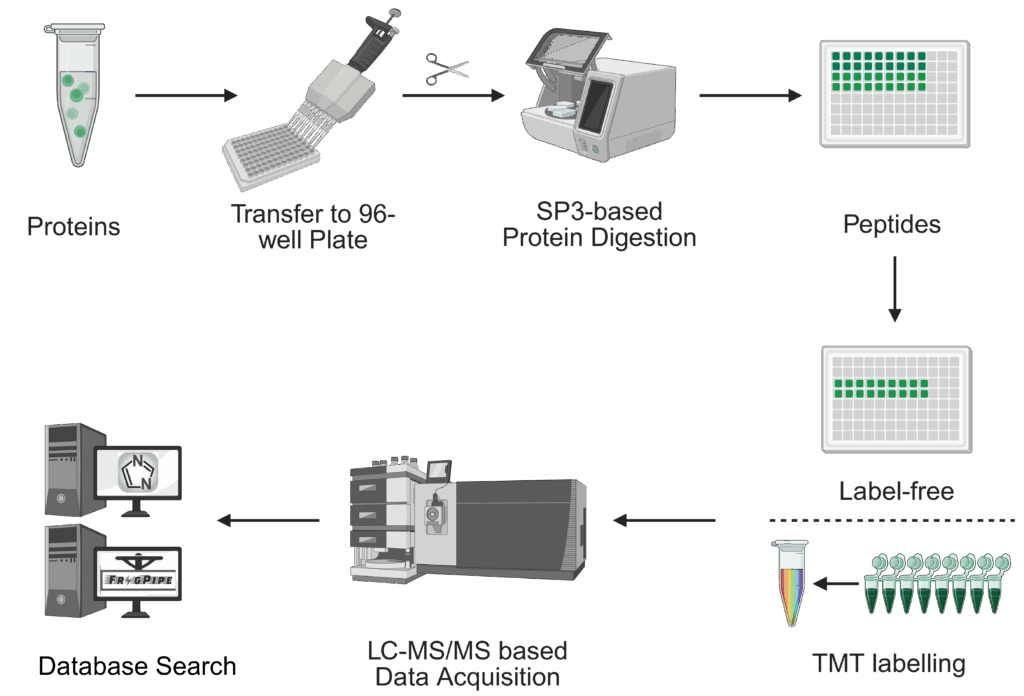
We provide support for relative protein quantification on:
Depending on the type and nature of the experiment, we use the following techniques for quantification:
We perform analysis of post-translational modifications (PTMs) either on level of purified proteins or – for selected PTMs – on a global scale.
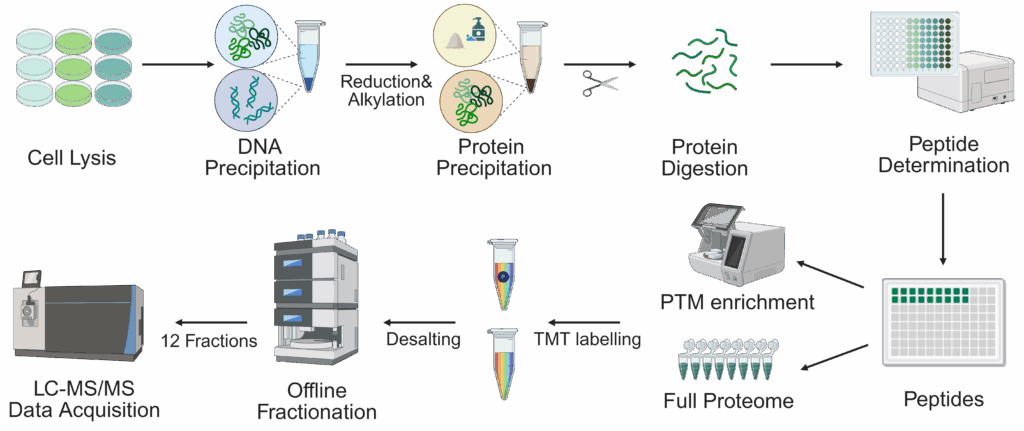
For analysis of PTMs on a global scale, we provide support for quantitative analysis of:
For analysis of PTMs from purified proteins, please provide as with your sample from an SDS-PAGE as well as information on the nature of the modification(s).
See here for more information.
We provide support in the analysis of protein-protein interactions of purified proteins and protein complexes using chemical crosslinking coupled to mass spectrometry (XL-MS).
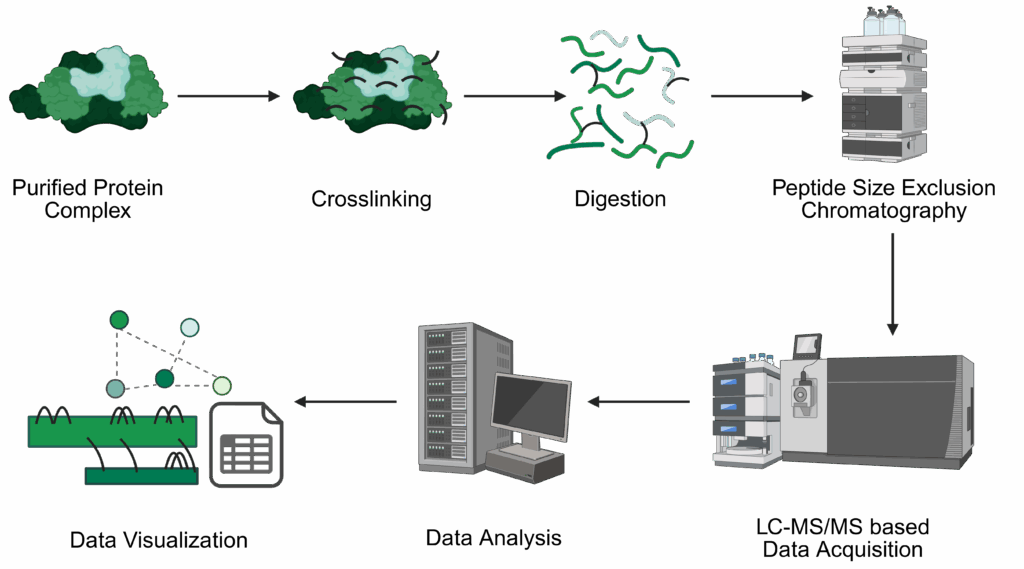
At the moment, we provide XL-MS using the following crosslinkers
For this analysis, you will perform the crosslinking in your lab using our provided protocol up to the quenching step, then send us the sample for further processing. This ensures optimal experimental conditions during the crosslinking step and preserves sample integrity.
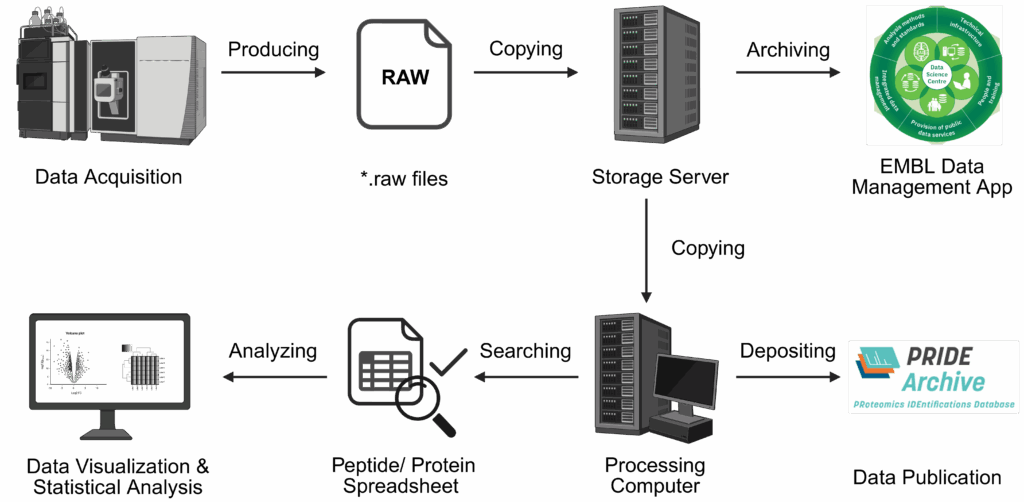
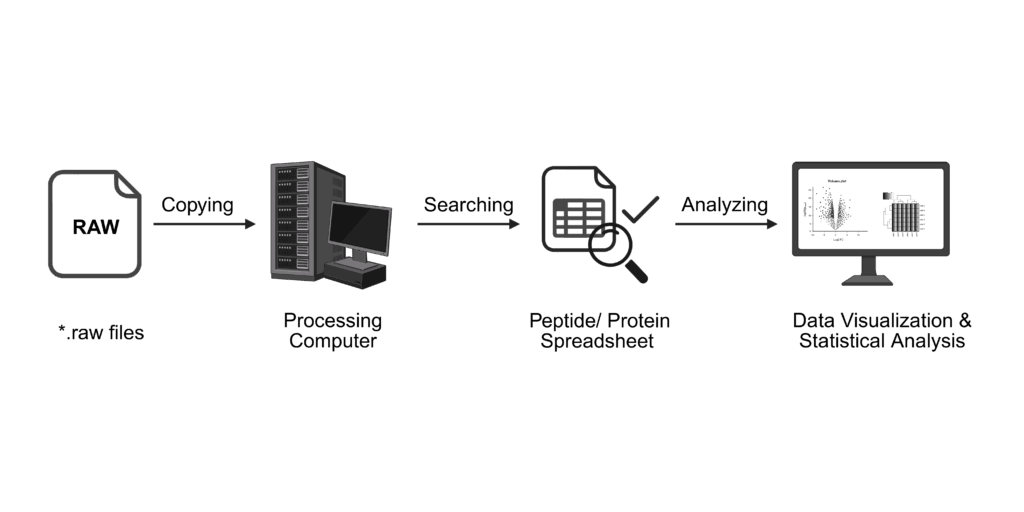
Our support includes data analysis of your samples. Depending on your workflow, your data will be searched and analysed accordingly.
For all sample types, our data analysis includes
For more details on the different analyses and their deliveries, please read the information for the respective service below.
You can find more information on our standard data analysis in our Github repository.
For protein identification, we will additionally provide sequence alignment plots and other descriptive visualizations. Data analysis for protein identification will be performed using MSFragger in the FragPipe suite using either a database provided by you or the respective Uniprot reference proteome.
For protein quantification, we provide a comprehensive data analysis of the proteomics experiment along with the complete R script to ensure full reproducibility.
Our statistical pipeline includes
Differential expression is assessed using moderated t-statistics from the limma package, and results are visualised through correlation plots and heat maps with clustering to highlight patterns in the data.
To support biological interpretation, we also provide a basic Gene Ontology enrichment analysis for standard model organisms.
For TMT, data analysis will be performed using MSFragger in the FragPipe suite using either a database provided by you or the respective Uniprot reference proteome.
For DIA, data analysis will be performed using DIA-NN using either a database provided by you or the respective Uniprot reference proteome.
For PTM analysis on a global scale, we provide a similar type of data analysis as for protein quantification data. Additionally, we include peptide-centric data and visualizations.
For PTM analysis from purified proteins, we will additionally provide sequence alignment plots and other descriptive visualizations.
For TMT and DDA, data analysis will be performed using MSFragger in the FragPipe suite using either a database provided by you or the respective Uniprot reference proteome.
For DIA, data analysis will be performed using DIA-NN using either a database provided by you or the respective Uniprot reference proteome.
For crosslinking, we provide spreadsheets containing all confidently identified crosslink peptides. This includes a datasheet that is ready for import to xiView where you can interactively investigate your data, visualize protein-protein interaction networks and map crosslinks on publicly available 3D structures.
Crosslink analysis will be performed using pLink3.

How to submit your samples at the PCF