Stronger together
Binding of transcription factors measured on individual DNA molecules in living cells
An important question in the field of gene regulation is how a limited set of transcription factors interpret the same genome in different ways to create over 200 cell types found in our bodies, all of which are defined by the expression of a unique set of genes. To achieve this, transcription factors have evolved cooperative mechanisms to enable precise control of gene expression. In a new study, members of the Krebs group in the Genome Biology Unit at EMBL Heidelberg have developed a method to directly visualise how transcription factors co-occupy DNA in living cells, and have used it to better understand their cooperativity mechanisms. “Our results improve our fundamental understanding of the genetic code that regulates expression of genes, and will be critical to interpret genetic differences between individuals and genetic alterations in diseases,” explains group leader Arnaud Krebs.
Transcription factors are proteins that bind to specific regulatory DNA sequences to control which genes are turned on or off. Our DNA is tightly packaged with proteins called histones, creating a complex of DNA and protein called chromatin. For transcription factors to activate regulatory regions of the DNA, histones typically need to be removed so the transcription factors can access their recognition sites. Transcription factors often work collectively to achieve this task – scientists speak of cooperativity. While this process is critical for the definition of a cell type during development, the underlying mechanisms for these collective actions are mostly unknown. “Therefore, we developed an experimental setup and combined it with a unique computational approach to capture cooperativity in action,” explains PhD student and lead author Can Sönmezer.
New method leads to new insights
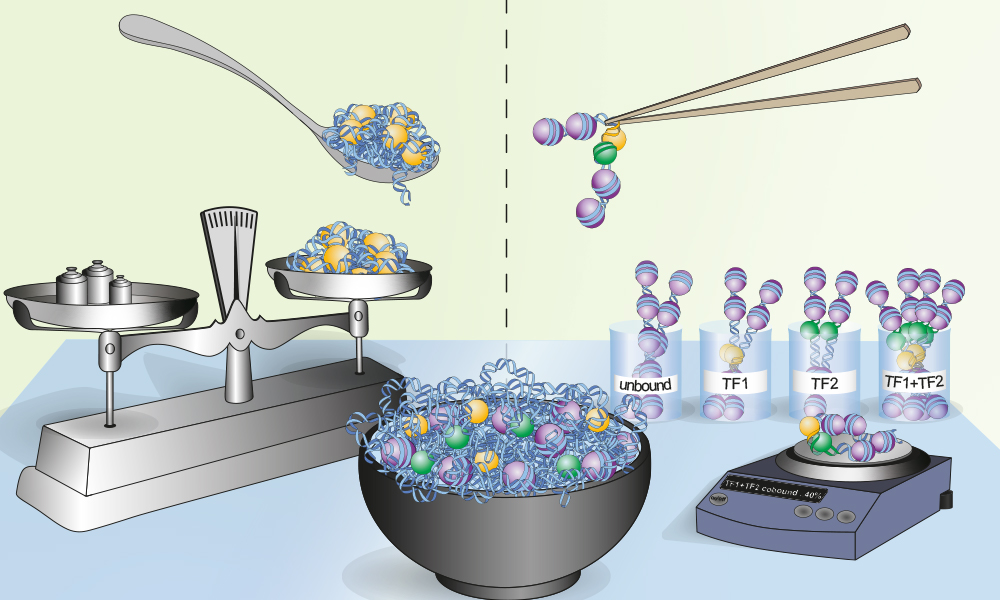
“Current technologies for studying transcription factor binding mostly rely on averaged information from thousands to millions of cells,” explains Krebs. “Although very informative, such bulk data cannot be used to understand whether multiple transcription factors co-bind the same DNA molecule in a living cell. Given that co-occupancy of transcription factors is hypothesised to be an important driver of their biological functions, we set out to overcome this limitation.”
The new approach by Krebs and his team members, known as single-molecule footprinting, can resolve the binding of multiple transcription factors on individual DNA molecules. This allows the researchers to determine if and where transcription factors co-bind their target regions. They discovered that the degree of co-occupancy is very variable throughout the genome, and mostly independent of which two factors are involved in a pair. Strikingly, co-occupancy of transcription factors scales with how densely a regulatory element is packaged in chromatin. This finding hints that the more difficult a region is to ‘unpackage’ and to activate, the more transcription factors depend on their partners to exert their functions. The resolution at which Krebs and his group measured transcription factor and nucleosome binding events is new to the field, making the study the first in which the co-occupancy of transcription factors has been measured at the single-molecule level genome-wide.
Open questions despite unprecedented details
While the study was able to answer some longstanding questions about transcription factor cooperativity, it also raised new ones. For instance, mathematical modelling of the co-occupancy data surprisingly revealed that the phenomenon they observed cannot simply be explained by an equilibrium between the binding of transcription factors and histones, as previously proposed. Their data suggests that recruitment of enzymes to remodel chromatin is likely to be involved in the process.
“Our work led to surprising findings, and new hypotheses that we are excited to test in the lab. This illustrates for me the importance of quantitative data modelling to understand a biological mechanism,” Krebs says.