Solving the nucleosome: twenty years on
Ahead of the EMBO conference The Nucleosome: From Atoms to Genomes, speaker Tim Richmond looks back on the work that revealed the high-resolution structure of the nucleosome
The DNA double helix is surely the most famous structure in molecular biology. But the familiar image of a linear DNA strand bears little resemblance to the elaborate organisation of DNA inside cells. Part of the reason for this high level of organisation is the need to pack around two metres of linear DNA into a cell’s nucleus – a space less than one hundred thousandth of a metre across. But that’s not the whole story. The three-dimensional arrangement of the DNA plays an important role in regulating gene activity, for example by making certain DNA regions more accessible to molecules that switch genes on or off. “All this organisation is important but the process of gene regulation is not well understood from a structural point of view,” says Richmond, a professor at ETH Zürich in Switzerland. “We’ve been asking questions about this for a long time but the answers have not been so easy to get at.”

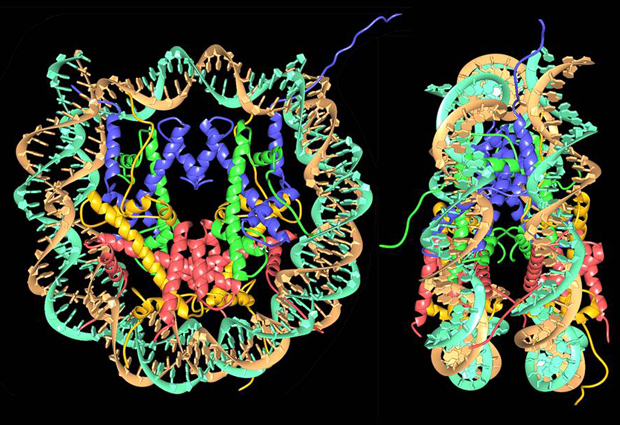
Starting from a linear DNA strand, the first level of organisation involves a structure called the nucleosome, in which DNA is wound for two turns around a cluster of eight proteins called histones. A single strand of DNA has many nucleosomes at regular intervals along its length – an arrangement that’s often compared to beads on a string. The string is then coiled up, causing the nucleosomes to pack more closely together, and there are additional levels of organisation involving other kinds of scaffolding proteins – compacting the structure still further – although these levels of structure have not yet been determined in detail.
In the early 1980s the structure of the nucleosome was still mysterious, although several research groups were working on the problem. “There were all kinds of models, but people really didn’t have a good idea of what it looked like,” says Richmond, who was then part of a group led by Aaron Klug at the MRC Laboratory of Molecular Biology in Cambridge. In 1984, Klug’s group published a structure of the nucleosome with a resolution of 7 Å (1 Å, or ångström, is one ten billionth of a metre – about the size of a typical atom). This confirmed the disc-like shape of the nucleosome and provided some additional information about the structure of the histones, but it still did not enable the structure to be determined at the atomic level.
Difficult phases
Klug’s group used a standard technique in structural biology: X-ray crystallography. This involves taking crystals of a specific molecule and exposing them to an intense beam of X-rays. The X-rays are scattered by the crystals, producing a pattern of bright and dark spots that can be recorded. By studying this pattern, it’s possible to work out the structure of the molecule. However, in all X-ray crystallography studies there’s a significant obstacle that has to be overcome: the phase problem.
X-rays, like light, are a form of electromagnetic wave. If you think of this wave as an undulating line with successive peaks and troughs, the phase describes which part of its cycle the wave is in currently: whether it’s near a peak, near a trough, or somewhere in between. In X-ray crystallography, it’s important to know the differences in phase between X-rays scattered by different parts of the crystal, because this provides vital extra information for determining the crystal structure. The reason this is described as a problem is that the phase information can’t be measured directly, by looking at the pattern of bright and dark spots. Various methods can be used to solve the phase problem: for the nucleosome, the researchers used a technique that involves incorporating heavy atoms – in this case, atoms of mercury – into the crystal structure. By comparing results from samples with and without these heavy atoms, it’s possible to infer the necessary phase information. “Another group was formulating models and trying to work out the phases from their model,” explains Richmond. “But we managed to solve the problem using physics. Incorporating heavy atoms is a standard method in X-ray crystallography but it was hard to achieve for such a large structure back then.”
Incorporating a compound containing mercury into the structure was one of the things Richmond worked on during his time in Cambridge. He subsequently moved to ETH Zürich, where he led a group of scientists who determined the structure of the nucleosome with a resolution of 2.8 Å. This high-resolution structure was published in Nature in 1997.
Structural refinements
Getting to the high-resolution structure involved many important steps made by Richmond’s group and others. To obtain high-quality data from X-ray crystallography, the sample needs to be arranged in well-ordered crystals where the molecules are regularly spaced and in near-identical configurations. One of the challenges in trying to do this using nucleosomes from a typical cell is that the DNA sequences around each nucleosome are not the same, and the stretches of DNA between nucleosomes can vary in length. An important step was therefore to genetically engineer DNA sequences of defined length in E. coli. “That made our crystallography images much clearer than before,” says Richmond.
It was also possible to improve the resolution by using strands of DNA with sequences that read as a palindrome. The reason for this is that the nucleosome contains eight histones of four different types arranged symmetrically in pairs. By using palindromic sequences of DNA, this symmetry can be preserved – approximately – when the DNA is wrapped around the histones, so that the sequence running along one side of the nucleosome is matched symmetrically by the sequence on the other side, producing clearer crystallography images.
I didn’t really feel euphoric. It was more a sense of relief
At the time of the Nature paper, several groups were racing to determine the nucleosome’s structure. “That creates a lot of pressure, and you have to have confidence that you’re going to get there first,” says Richmond. When his group published their high-resolution structure in 1997, it was eighteen years since he’d first started working on the problem. Once they had the structure, he explains, “I didn’t really feel euphoric. It was more a sense of relief. You don’t feel the accomplishment at first because you’ve been with it for so long.” But he’s clear about the importance of taking time to recognise what you’ve achieved. “In science you’re always moving on to the next step. At some point you have to take a few days off and look back and see that you actually did accomplish something, because that really helps you the next time it gets tough.”
Three-dimensional code
Richmond’s work now focuses on the higher levels of organisation of DNA, investigating the way nucleosomes are packed together in chromatin – the material that makes up our chromosomes – and how this influences gene regulation. “There’s the linear genetic code, but then the three-dimensional organisation of the DNA is like another layer of code that largely determines how regulation works,” Richmond says. “Everything goes back to that three-dimensional structure.” Understanding how this arrangement of our DNA affects the activity of our genes is an incredibly complex task, but progress is being made. “Over the past year or so we’ve had some success,” he explains. “I hope to be in a position to discuss that at the conference.”