Current research
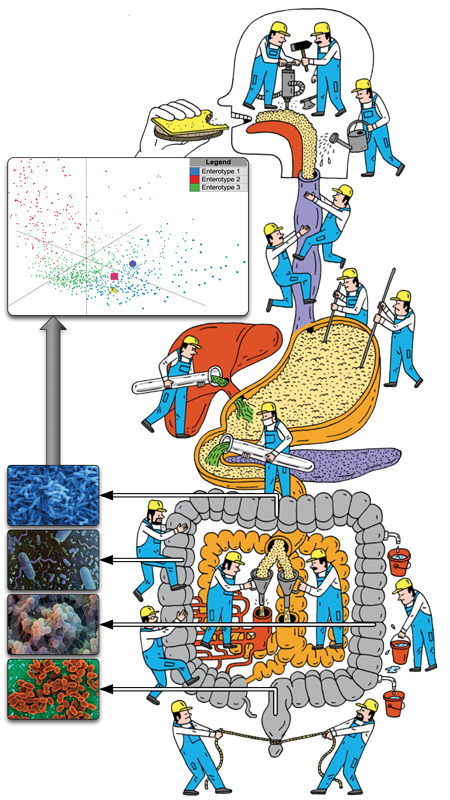
The group works across spatial scales, from genes, proteins, and small molecules over molecular and cellular networks to microbial communities. We focus on the computational analysis of microbiome data from both the human gut and the environment. By integrating large-scale datasets of public and in-house metagenomic data, we aim to find diagnostic and prognostic biomarkers of diseases, and look for patterns that only become apparent at a global scale, such as enterotypes or variations in microbial load.
Our goal is to achieve a functional understanding of microbial community interactions, both among microbes and with their environment. This includes tracing horizontal gene transfer (e.g. by mobile elements) and mapping functional traits at strain level (approximated by functional modules) across phylogeny and habitats. Additionally, we work to identify microbial early response biomarkers for man-made pollutants, such as medication, pesticides and other toxins. In collaboration with other groups at EMBL, we aim to establish interaction maps between chemical compounds and microbes, both individually and in communities, using advanced multi-omics approaches. Towards planetary health, we will establish comprehensive maps of species and gene fluxes on earth to understand, for example, the spread of antimicrobial resistance, where horizontal gene transfer plays a major role. As part of this effort, we helped initiate and participated in the TREC Expedition along Europe’s coastlines, and are now analyzing the gathered data.
Previous research
We often work in emerging research areas, balancing method development with biological discoveries. Notable past frontier research projects include participation in the Human Genome Project (Lander et al., Nature 2001), foundational studies of protein interaction networks (von Mering et al., Nature 2002), new concepts for the construction of a highly resolved tree of life (Ciccarelli et al., Science 2006), as well as exploration of drug–target interactions using global human ‘read-outs’ such as side-effects (Campillos et al., Science 2008).
In the microbiome field, we conducted with collaborators the first comparative metagenomic studies (Tringe et al., Science 2005). We have been using metagenomics to uncover principles governing microbial communities in healthy individuals and patients, for example playing a key role in the MetaHIT consortium which was transformational in the gut microbiome field (Qin et al., Nature 2010). We identified three main ‘enterotypes’, i.e. gut microbial community compositions (Arumugam et al., Nature 2011), and showed that each human appears to carry individual strains (Schloissnig et al., Nature 2013). We used strain tracking to quantify strain dynamics following fecal microbiota transplantation (Schmidt et al., Nature Medicine, 2022).
Towards practical applications, we have identified microbial markers for a number of diseases, such as obesity (Le Chatelier et al., Nature 2013), colon cancer (Zeller et al., Mol Sys Biol 2014; Wirbel et al., Nature Med 2019) and pancreatic cancer (Kartal et al., Gut 2022), applicable to diagnostics; and demonstrated the impact of drugs on the gut microbiome (e.g. Forslund et al., Nature 2015; Maier et al., Nature 2018), which could influence individualized treatment strategies.
Studying the microbiome of global environments, we participated the Tara Oceans Consortium, which uses the schooner Tara for expeditions (e.g. Bork et al., Science 2015, and references therein) and have been working on soils by global sampling (Bahram et al., Nature 2018). Integrating metagenome data from all environments, we created a large gene catalog to study the biogeography of prokaryotic genes (Coelho et al., Nature, 2022).
Databases and services
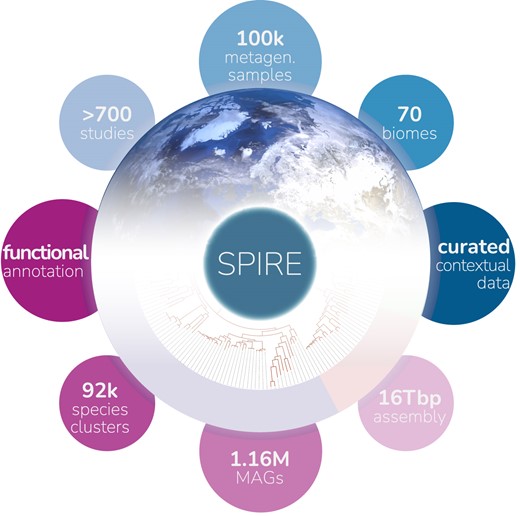
We are engaged in tool and resource development to support our work, ranging from functional annotation (SMART, EggNOG, proGenomes), via visualisation (iTOL), to metagenomics profiling and global gene cataloguing (GMGC) and metagenome-assembled genomes and associated annotations (SPIRE). For a complete list including the associated publications, please refer to our group homepage.