Dealing with subworkflows in Snakemake

Introduction
Subdividing your pipeline into subworkflows is a powerful mean to achieve complex analysis. It also enables to avoid code repetition. Typically, if one has to
analyse different types of data such as ChIP-seq and RNA-seq, and then perform multi-omics data integration, it becomes handy to create 2 subworflows (ChIPseq, RNAseq) and
connect the different types of data in a MultiOmics workflow.
In this tutorial, we will use toy examples that will lead to creating the subworkflows ChIPSeq and RNAseq. These subworkflows will be connected into a third one. Check the previous posts on “getting started with snakemake“, “creating a profile“, and
“submitting jobs to a cluster” if you are not familiar with these concepts.
We start by preparing the folder structure, testing the subworkflows one after the other, and finish by coordinating the merging of the data into one Snakefile.
Preparing the files
Run the following script to create the folder structure:
#!/usr/bin/bash
## Create the project folder
mkdir demo-subworkflows
cd demo-subworkflows
## create the folder and the file for the conda environment
mkdir envs
touch envs/environment.yaml
## create the folder and the file for the profile
mkdir profile
touch profile/config.yaml
## create the Snakefile that will connect the subworkflows ChIPSeq and RNASeq
touch Snakefile
## Create the ChIPSeq workflow files
mkdir ChIPSeq
touch ChIPSeq/Snakefile-ChIPSeq
mkdir ChIPSeq/profileChIPSeq
touch ChIPSeq/profileChIPSeq/config.yaml
touch ChIPSeq/profileChIPSeq/status-sacct.sh
mkdir ChIPSeq/inputs
touch ChIPSeq/inputs/exp1.fastq # This is a toy example, the file is empty
touch ChIPSeq/inputs/exp2.fastq # This is a toy example, the file is empty
mkdir ChIPSeq/external_rules
touch ChIPSeq/external_rules/mockrule.smk
touch ChIPSeq/external_rules/callscript.smk
mkdir ChIPSeq/scripts
touch ChIPSeq/scripts/mockscript.R
## Create the RNASeq workflow files
mkdir RNASeq
touch RNASeq/Snakefile-RNASeq
mkdir RNASeq/profileRNASeq
touch RNASeq/profileRNASeq/config.yaml
touch RNASeq/profileRNASeq/status-sacct.sh
mkdir RNASeq/inputs
touch RNASeq/inputs/exp1.fastq # This is a toy example, the file is empty
touch RNASeq/inputs/exp2.fastq # This is a toy example, the file is empty
mkdir RNASeq/external_rules
touch RNASeq/external_rules/mockrule.smk
touch RNASeq/external_rules/callscript.smk
mkdir RNASeq/scripts
touch RNASeq/scripts/mockscript.R
## Create the MultiOmics workflow files
mkdir MultiOmics
touch MultiOmics/Snakefile-MultiOmics
mkdir MultiOmics/profileMultiOmics
touch MultiOmics/profileMultiOmics/config.yaml
mkdir MultiOmics/external_rules
touch MultiOmics/external_rules/mockrule.smk
For more information about the YAML format see here. For the next step, you need to have
conda installed on your computer. Copy the following content to envs/environment.yaml:
channels:
- bioconda
dependencies:
- snakemake=7.8.2Then execute the following command to create a conda environment containing snakemake v7.8.2:
#!/usr/bin/bash
conda env create -p envs/smake --file envs/environment.yamlIf you get stuck with the installation at Solving environment, follow these steps:
#!/usr/bin/bash
conda create -p envs/smake python=3.9.13
conda activate envs/smake
conda install -c bioconda snakemake=7.8.2
You can verify that the correct version of snakemake has been installed with:
#!/usr/bin/bash
## If you did not already activate the environment: conda activate envs/smake
snakemake --version
conda deactivate
In the next section, we are going to build and test the ChIPSeq subworkflow.
The ChIPSeq subworkflow
If you are not familiar with the different steps to process ChIP-seq data, read this tutorial
by the galaxy training network.
Copy the following content into ChIPSeq/Snakefile-ChIPSeq:
# Load pipeline: snakemake --profile profileChIPSeq/ -n
onstart:
print("##### ChIPSeq workflow #####\n")
print("\t Creating jobs output subfolders...\n")
shell("mkdir -p jobs/mockAlignment")
shell("mkdir -p jobs/mockpeakDetection")
#########
# Variable definition
########
FILESNAMES=["exp1", "exp2"]
#########
# Rules
########
rule all:
input:
expand("results/alignment/{sampleName}.bam", sampleName=FILESNAMES),
expand("results/peaks/{sampleName}.peaks", sampleName=FILESNAMES)
include: "external_rules/mockrule.smk"
include: "external_rules/callscript.smk"The rule to copy in ChIPSeq/external_rules/mockrule.smk is:
rule mockAlignment:
input:
"inputs/{sampleName}.fastq"
output:
"results/alignment/{sampleName}.bam"
threads: 1
shell:
"""
cat {input} > {output}
sleep 10s
"""In a real context, the shell section of the code above will be more complex. It would contain instructions to align your fastq files to a reference genome. Your fastq files would be in gz format and would have been trimmed. Above is just a toy example that creates an empty file.
The second rule calls an R script from which a mock peak detection is performed. In a real ChIPSeq pipeline, more steps would be required. Important: Note that in the code below, the path to the script is relative to the position of ChIPSeq/external_rules/callscript.smk. Add this rule toChIPSeq/external_rules/callscript.smk:
rule mockpeakDetection:
input:
"results/alignment/{sampleName}.bam"
output:
"results/peaks/{sampleName}.peaks"
threads: 1
script:
"../scripts/mockscript.R"The input of the rule mockpeakDetection is the output of the rule mockAlignment. It is said that mockPeakDetection depends on mockAlignment. In other words, in order to perform the peak detection (on the bam files), you first need to generate the bam files by performing the alignment of the fastq files. Let’s visualize the directed acyclic graph of the jobs. It gives a representation of the relationships between the rules of a workflow:
#!/usr/bin/sh
conda activate envs/smake
cd ChIPSeq
snakemake --snakefile Snakefile-ChIPSeq --cores 1 -n --rulegraph | dot -Tpng > dag.pngYou should obtain the following graph:
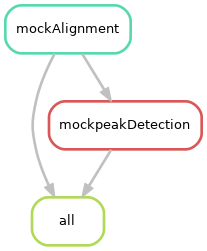
The scripts/mockscript.R must contain:
outputFile <- snakemake@output[[1]]
message("Writing to ", outputFile)
towrite <- "hello"
write(towrite, file=outputFile, ncolumns=1)Before running the workflow, we need to create the profile in order to submit the jobs to a cluster. For more information, see the links at the top of this page. If you are not using slurm, you will need to modify the code below. I also provide below the command to run the workflow in local. Copy the following content in profileChIPSeq/config.yaml:
---
snakefile: Snakefile-ChIPSeq
latency-wait: 60
reason: True
show-failed-logs: True
keep-going: True
printshellcmds: True
# Cluster submission
jobname: "{rule}.{jobid}" # Provide a custom name for the jobscript that is submitted to the cluster.
max-jobs-per-second: 1 #Maximal number of cluster/drmaa jobs per second, default is 10, fractions allowed.
max-status-checks-per-second: 10 #Maximal number of job status checks per second, default is 10
jobs: 400 #Use at most N CPU cluster/cloud jobs in parallel.
cluster: "sbatch --output=\"jobs/{rule}/slurm_%x_%j.out\" --error=\"jobs/{rule}/slurm_%x_%j.log\" --mem={resources.mem_mb} --time={resources.runtime} --parsable"
cluster-status: "./profileChIPSeq/status-sacct.sh" # Use to handle timeout exception, do not forget to chmod +x
# Job resources
set-resources:
- mockAlignment:mem_mb=1000
- mockAlignment:runtime=00:03:00
- mockpeakDetection:mem_mb=1000
- mockpeakDetection:runtime=00:03:00
# For some reasons time needs quotes to be read by snakemake
default-resources:
- mem_mb=500
- runtime="00:01:00"
# Define the number of threads used by rules
set-threads:
- mockAlignment=1
- mockpeakDetection=1Add profileChIPSeq/status-sacct.sh:
#!/usr/bin/env bash
# Check status of Slurm job
jobid="$1"
if [[ "$jobid" == Submitted ]]
then
echo smk-simple-slurm: Invalid job ID: "$jobid" >&2
echo smk-simple-slurm: Did you remember to add the flag --parsable to your sbatch call? >&2
exit 1
fi
output=`sacct -j "$jobid" --format State --noheader | head -n 1 | awk '{print $1}'`
if [[ $output =~ ^(COMPLETED).* ]]
then
echo success
elif [[ $output =~ ^(RUNNING|PENDING|COMPLETING|CONFIGURING|SUSPENDED).* ]]
then
echo running
else
echo failed
fiNow run the pipeline, either using the profile to submit to the cluster, or the second command to run it locally. You can also remove everything from the # cluster submission line in the profile to run the workflow in local and using the first command:
#!/usr/bin/bash
## Make `profile/status-sacct.sh` executable if you submit to a cluster
chmod +x profileChIPSeq/status-sacct.sh
## Submitting to the cluster (or in local if you deleted the relevant lines in the profile)
snakemake --profile profileChIPSeq/
## OR
## Running in local
snakemake --snakefile Snakefile-ChIPSeq --cores=1You should see:
Building DAG of jobs...
Using shell: /usr/bin/bash
Provided cluster nodes: 400
Job stats:
job count min threads max threads
----------------- ------- ------------- -------------
all 1 1 1
mockAlignment 2 1 1
mockpeakDetection 2 1 1
total 5 1 1
##### ChIPSeq workflow #####
Creating jobs output subfolders...
mkdir -p jobs/mockAlignment
mkdir -p jobs/mockpeakDetection
Select jobs to execute...
[Thu Mar 31 16:31:49 2022]
rule mockAlignment:
input: inputs/exp1.fastq
output: results/alignment/exp1.bam
jobid: 1
reason: Missing output files: results/alignment/exp1.bam
wildcards: sampleName=exp1
resources: mem_mb=1000, disk_mb=1000, tmpdir=/tmp, runtime=00:03:00
cat inputs/exp1.fastq > results/alignment/exp1.bam
sleep 10s
Submitted job 1 with external jobid '37058084'.
[Thu Mar 31 16:31:49 2022]
rule mockAlignment:
input: inputs/exp2.fastq
output: results/alignment/exp2.bam
jobid: 2
reason: Missing output files: results/alignment/exp2.bam
wildcards: sampleName=exp2
resources: mem_mb=1000, disk_mb=1000, tmpdir=/tmp, runtime=00:03:00
cat inputs/exp2.fastq > results/alignment/exp2.bam
sleep 10s
Submitted job 2 with external jobid '37058085'.
Waiting at most 60 seconds for missing files.
[Thu Mar 31 16:32:40 2022]
Finished job 1.
1 of 5 steps (20%) done
Select jobs to execute...
[Thu Mar 31 16:32:40 2022]
rule mockpeakDetection:
input: results/alignment/exp1.bam
output: results/peaks/exp1.peaks
jobid: 3
reason: Missing output files: results/peaks/exp1.peaks; Input files updated by another job: results/alignment/exp1.bam
wildcards: sampleName=exp1
resources: mem_mb=1000, disk_mb=1000, tmpdir=/tmp, runtime=00:03:00
Submitted job 3 with external jobid '37058227'.
[Thu Mar 31 16:32:40 2022]
Finished job 2.
2 of 5 steps (40%) done
Select jobs to execute...
[Thu Mar 31 16:32:40 2022]
rule mockpeakDetection:
input: results/alignment/exp2.bam
output: results/peaks/exp2.peaks
jobid: 4
reason: Missing output files: results/peaks/exp2.peaks; Input files updated by another job: results/alignment/exp2.bam
wildcards: sampleName=exp2
resources: mem_mb=1000, disk_mb=1000, tmpdir=/tmp, runtime=00:03:00
Submitted job 4 with external jobid '37058228'.
Waiting at most 60 seconds for missing files.
[Thu Mar 31 16:33:10 2022]
Finished job 3.
3 of 5 steps (60%) done
[Thu Mar 31 16:33:10 2022]
Finished job 4.
4 of 5 steps (80%) done
Select jobs to execute...
[Thu Mar 31 16:33:10 2022]
localrule all:
input: results/alignment/exp1.bam, results/alignment/exp2.bam, results/peaks/exp1.peaks, results/peaks/exp2.peaks
jobid: 0
reason: Input files updated by another job: results/peaks/exp2.peaks, results/peaks/exp1.peaks, results/alignment/exp2.bam, results/alignment/exp1.bam
resources: mem_mb=500, disk_mb=1000, tmpdir=/tmp, runtime=00:01:00
[Thu Mar 31 16:33:10 2022]
Finished job 0.
5 of 5 steps (100%) done
Complete log: demo-subworkflows/ChIPSeq/.snakemake/log/2022-03-31T163148.828799.snakemake.logLet’s verify that ChIPSeq/external_rules/mockrule.smk created correctly the bam files:
#!/usr/bin/bash
## It should print: results/alignment/exp1.bam results/alignment/exp2.bam
ls results/alignment/*Now check that the “peak detection” created the files correctly. Each file should contain “hello” that was added by scripts/mockscript.R:
#!/usr/bin/bash
## Checking that the files were created
## It should print: results/peaks/exp1.peaks results/peaks/exp2.peaks
ls results/peaks/*
## Checking the content of each file, You should see the string "hello" in each file.
more results/peaks/*Change the current shell location to RNASeq:
#!/usr/bin/bash
cd ../RNASeqThe RNA-Seq subworkflow
If you are not familiar with the different steps to process RNA-seq data, read this tutorial
by the galaxy training network. Below we will create two toy rules mockFeatureCounts and mockDifferentialAnalysis that will be contained in external_rules/mockrule.smk andexternal_rules/callscript.smk.
Copy the following content into Snakefile-RNASeq:
# Load pipeline: snakemake --profile profileRNASeq/ -n
onstart:
print("##### RNASeq workflow #####\n")
print("\t Creating jobs output subfolders...\n")
shell("mkdir -p jobs/mockFeatureCounts")
shell("mkdir -p jobs/mockDifferentialAnalysis")
#########
# Variable definition
########
FILESNAMES=["exp1", "exp2"]
#########
# Rules
########
rule all:
input:
expand("results/featureCounts/{sampleName}.txt", sampleName=FILESNAMES),
expand("results/differentialAnalysis/{sampleName}.csv", sampleName=FILESNAMES)
include: "external_rules/mockrule.smk"
include: "external_rules/callscript.smk"The rule to copy in external_rules/mockrule.smk is:
rule mockFeatureCounts:
input:
"inputs/{sampleName}.fastq"
output:
"results/featureCounts/{sampleName}.txt"
threads: 1
shell:
"""
cat {input} > {output}
sleep 10s
"""The rule to copy in external_rules/callscript.smk is:
rule mockDifferentialAnalysis:
input:
"results/featureCounts/{sampleName}.txt"
output:
"results/differentialAnalysis/{sampleName}.csv"
threads: 1
script:
"../scripts/mockscript.R"Add the following content to scripts/mockscript.R:
outputFile <- snakemake@output[[1]]
message("Writing to ", outputFile)
towrite <- "This is a differential gene expression analysis normally performed with a package such as deseq2"
write(towrite, file=outputFile, ncolumns=1)As we did in the previous section, create profileRNASeq/config.yaml with the following content:
---
snakefile: Snakefile-RNASeq
latency-wait: 60
reason: True
show-failed-logs: True
keep-going: True
printshellcmds: True
# Cluster submission
jobname: "{rule}.{jobid}" # Provide a custom name for the jobscript that is submitted to the cluster.
max-jobs-per-second: 1 #Maximal number of cluster/drmaa jobs per second, default is 10, fractions allowed.
max-status-checks-per-second: 10 #Maximal number of job status checks per second, default is 10
jobs: 400 #Use at most N CPU cluster/cloud jobs in parallel.
cluster: "sbatch --output=\"jobs/{rule}/slurm_%x_%j.out\" --error=\"jobs/{rule}/slurm_%x_%j.log\" --mem={resources.mem_mb} --time={resources.runtime} --parsable"
cluster-status: "./profileRNASeq/status-sacct.sh" # Use to handle timeout exception, do not forget to chmod +x
# Job resources
set-resources:
- mockFeatureCounts:mem_mb=1000
- mockFeatureCounts:runtime=00:03:00
- mockDifferentialAnalysis:mem_mb=1000
- mockDifferentialAnalysis:runtime=00:03:00
# For some reasons time needs quotes to be read by snakemake
default-resources:
- mem_mb=500
- runtime="00:01:00"
# Define the number of threads used by rules
set-threads:
- mockFeatureCounts=1
- mockDifferentialAnalysis=1Exactly as before, add profileRNASeq/status-sacct.sh:
#!/usr/bin/env bash
# Check status of Slurm job
jobid="$1"
if [[ "$jobid" == Submitted ]]
then
echo smk-simple-slurm: Invalid job ID: "$jobid" >&2
echo smk-simple-slurm: Did you remember to add the flag --parsable to your sbatch call? >&2
exit 1
fi
output=`sacct -j "$jobid" --format State --noheader | head -n 1 | awk '{print $1}'`
if [[ $output =~ ^(COMPLETED).* ]]
then
echo success
elif [[ $output =~ ^(RUNNING|PENDING|COMPLETING|CONFIGURING|SUSPENDED).* ]]
then
echo running
else
echo failed
fiMake it executable and run the workflow either on the cluster or locally:
#!/usr/bin/bash
## Make `profile/status-sacct.sh` executable if you submit to a cluster
chmod +x profileRNASeq/status-sacct.sh
## Submitting to the cluster (or in local if you deleted the relevant lines in the profile)
snakemake --profile profileRNASeq/
## OR
## Running in local without profile
snakemake --snakefile Snakefile-RNASeq --cores=1
You should see in your terminal:
Building DAG of jobs...
Using shell: /usr/bin/bash
Provided cluster nodes: 400
Job stats:
job count min threads max threads
------------------------ ------- ------------- -------------
all 1 1 1
mockDifferentialAnalysis 2 1 1
mockFeatureCounts 2 1 1
total 5 1 1
##### RNASeq workflow #####
Creating jobs output subfolders...
mkdir -p jobs/mockFeatureCounts
mkdir -p jobs/mockDifferentialAnalysis
Select jobs to execute...
[Thu Jun 16 16:06:27 2022]
rule mockFeatureCounts:
input: inputs/exp1.fastq
output: results/featureCounts/exp1.txt
jobid: 1
reason: Missing output files: results/featureCounts/exp1.txt
wildcards: sampleName=exp1
resources: mem_mb=1000, disk_mb=1000, tmpdir=/tmp, runtime=00:03:00
cat inputs/exp1.fastq > results/featureCounts/exp1.txt
sleep 10s
Submitted job 1 with external jobid '42024859'.
[Thu Jun 16 16:06:27 2022]
rule mockFeatureCounts:
input: inputs/exp2.fastq
output: results/featureCounts/exp2.txt
jobid: 2
reason: Missing output files: results/featureCounts/exp2.txt
wildcards: sampleName=exp2
resources: mem_mb=1000, disk_mb=1000, tmpdir=/tmp, runtime=00:03:00
cat inputs/exp2.fastq > results/featureCounts/exp2.txt
sleep 10s
Submitted job 2 with external jobid '42024860'.
[Thu Jun 16 16:06:57 2022]
Finished job 1.
1 of 5 steps (20%) done
Select jobs to execute...
[Thu Jun 16 16:06:57 2022]
rule mockDifferentialAnalysis:
input: results/featureCounts/exp1.txt
output: results/differentialAnalysis/exp1.csv
jobid: 3
reason: Missing output files: results/differentialAnalysis/exp1.csv; Input files updated by another job: results/featureCounts/exp1.txt
wildcards: sampleName=exp1
resources: mem_mb=1000, disk_mb=1000, tmpdir=/tmp, runtime=00:03:00
Submitted job 3 with external jobid '42024877'.
[Thu Jun 16 16:06:57 2022]
Finished job 2.
2 of 5 steps (40%) done
Select jobs to execute...
[Thu Jun 16 16:06:57 2022]
rule mockDifferentialAnalysis:
input: results/featureCounts/exp2.txt
output: results/differentialAnalysis/exp2.csv
jobid: 4
reason: Missing output files: results/differentialAnalysis/exp2.csv; Input files updated by another job: results/featureCounts/exp2.txt
wildcards: sampleName=exp2
resources: mem_mb=1000, disk_mb=1000, tmpdir=/tmp, runtime=00:03:00
Submitted job 4 with external jobid '42024878'.
Waiting at most 60 seconds for missing files.
[Thu Jun 16 16:07:28 2022]
Finished job 3.
3 of 5 steps (60%) done
[Thu Jun 16 16:07:28 2022]
Finished job 4.
4 of 5 steps (80%) done
Select jobs to execute...
[Thu Jun 16 16:07:28 2022]
localrule all:
input: results/featureCounts/exp1.txt, results/featureCounts/exp2.txt, results/differentialAnalysis/exp1.csv, results/differentialAnalysis/exp2.csv
jobid: 0
reason: Input files updated by another job: results/differentialAnalysis/exp2.csv, results/differentialAnalysis/exp1.csv, results/featureCounts/exp2.txt, results/featureCounts/exp1.txt
resources: mem_mb=500, disk_mb=1000, tmpdir=/tmp, runtime=00:01:00
[Thu Jun 16 16:07:28 2022]
Finished job 0.
5 of 5 steps (100%) done
Complete log: .snakemake/log/2022-06-16T160626.178736.snakemake.logLet’s verify that RNASeq/external_rules/mockrule.smk created correctly the featureCounts files:
#!/usr/bin/bash
## It should print: results/featureCounts/exp1.txt results/featureCounts/exp2.txt
ls results/featureCounts/*Now check that the “differential analysis” created the files correctly. Each file should contain the sentence “This is a differential gene expression analysis normally performed with a package such as deseq2” that was added by scripts/mockscript.R:
#!/usr/bin/bash
## Checking that the files were created
## It should print: results/differentialAnalysis/exp1.csv results/differentialAnalysis/exp2.csv
ls results/differentialAnalysis/*
## Checking the content of each file
more results/differentialAnalysis/*Go to the Multiomics folder of the project:
#!/usr/bin/bash
cd ../Multiomics
Connecting the ChIP-Seq and RNA-Seq subworkflow
Connecting the subworkflows in one Snakefile enables us to run them in one call but also to develop extra rules on top of them. One could thus build re-usable blocks that can be plugged into new projects and pipelines. We first run the connecting workflow locally. We will then see how to modify the files to run it on an HPC.
Connecting subworkflows locally
With Snakemake 6.0 and later, it is possible to define external workflows as modules, from which rules can be used by explicitly “importing” them. First, make sure that the user is using a correct version of snakemake by enforcing a minimum version (keep reading before adding to Snakefile-MultiOmics):
from snakemake.utils import min_version
min_version("6.0.0")
onstart:
print("##### ChIP-seq and RNA-seq workflows #####\n")
print("\t Reading samples and metadata....\n")Import the ChIPSeq and RNASeq subworkflows as modules (keep reading before adding):
from snakemake.utils import min_version
min_version("6.0.0")
onstart:
print("##### ChIP-seq and RNA-seq workflows #####\n")
print("\t Reading samples and metadata....\n")
module RNASeq:
snakefile:
"../RNASeq/Snakefile-RNASeq"
## This instruction should come right after the module
use rule * from RNASeq as RNASeq_*
module ChIPSeq:
snakefile:
"../ChIPSeq/Snakefile-ChIPSeq"
## This instruction should come right after the module
use rule * from ChIPSeq as ChIPSeq_*Let’s add the rule all (you can now copy the content to Snakefile-MultiOmics):
from snakemake.utils import min_version
min_version("6.0.0")
onstart:
print("##### ChIP-seq and RNA-seq workflows #####\n")
print("\t Reading samples and metadata....\n")
module RNASeq:
snakefile:
"../RNASeq/Snakefile-RNASeq"
## This instruction should come right after the module
use rule * from RNASeq as RNASeq_*
module ChIPSeq:
snakefile:
"../ChIPSeq/Snakefile-ChIPSeq"
## This instruction should come right after the module
use rule * from ChIPSeq as ChIPSeq_*
rule all:
input:
rules.RNASeq_all.input,
rules.ChIPSeq_all.inputNow try a dry-run:
#!/usr/bin/bash
snakemake --snakefile Snakefile-MultiOmics --cores 1 -n
You should get:
Building DAG of jobs...
MissingInputException in line 1 of /g/romebioinfo/tmp/demo-subworkflows/RNASeq/external_rules/mockrule.smk:
Missing input files for rule RNASeq_mockFeatureCounts:
output: results/featureCounts/exp1.txt
wildcards: sampleName=exp1
affected files:
inputs/exp1.fastq
Snakemake indicates that it cannot find the file inputs/exp1.fastq that corresponds to the input of the rule RNASeq/external_rules/mockrule.smk. Indeed, since we are currently in the Multiomics folder, the path is not correct. In order for snakemake to find the correct path, we must use the directive prefix. The string given to prefix will be added to every file path built by the snakemake process. Modify Snakefile-MultiOmics as follows:
from snakemake.utils import min_version
min_version("6.0.0")
onstart:
print("##### ChIP-seq and RNA-seq workflows #####\n")
print("\t Reading samples and metadata....\n")
module RNASeq:
prefix: "../RNASeq"
snakefile:
"../RNASeq/Snakefile-RNASeq"
## This instruction should come right after the module
use rule * from RNASeq as RNASeq_*
module ChIPSeq:
prefix: "../ChIPSeq"
snakefile:
"../ChIPSeq/Snakefile-ChIPSeq"
## This instruction should come right after the module
use rule * from ChIPSeq as ChIPSeq_*
rule all:
input:
rules.RNASeq_all.input,
rules.ChIPSeq_all.inputTry again a dry-run:
#!/usr/bin/bash
snakemake --snakefile Snakefile-MultiOmics --cores 1 -n
You should get:
Building DAG of jobs...
Nothing to be done (all requested files are present and up to date).The call to snakemake is now working. But since we already run the subworkflows ChIPSeq and RNASeq nothing has to be done. Delete the jobs and resultsfolders of each subworkflows and perform a dry-run:
#!/usr/bin/bash
rm -r ../RNASeq/jobs
rm -r ../RNASeq/results
rm -r ../ChIPSeq/jobs
rm -r ../ChIPSeq/results
snakemake --snakefile Snakefile-MultiOmics --cores 1 -n
You should get:
Building DAG of jobs...
Job stats:
job count min threads max threads
------------------------------- ------- ------------- -------------
RNASeq_all 1 1 1
RNASeq_mockDifferentialAnalysis 2 1 1
RNASeq_mockFeatureCounts 2 1 1
total 5 1 1
[Wed Jul 13 18:19:50 2022]
rule RNASeq_mockFeatureCounts:
input: ../RNASeq/inputs/exp2.fastq
output: ../RNASeq/results/featureCounts/exp2.txt
jobid: 2
reason: Missing output files: ../RNASeq/results/featureCounts/exp2.txt
wildcards: sampleName=exp2
resources: tmpdir=/tmp
[Wed Jul 13 18:19:50 2022]
rule RNASeq_mockFeatureCounts:
input: ../RNASeq/inputs/exp1.fastq
output: ../RNASeq/results/featureCounts/exp1.txt
jobid: 1
reason: Missing output files: ../RNASeq/results/featureCounts/exp1.txt
wildcards: sampleName=exp1
resources: tmpdir=/tmp
[Wed Jul 13 18:19:50 2022]
rule RNASeq_mockDifferentialAnalysis:
input: ../RNASeq/results/featureCounts/exp2.txt
output: ../RNASeq/results/differentialAnalysis/exp2.csv
jobid: 4
reason: Missing output files: ../RNASeq/results/differentialAnalysis/exp2.csv; Input files updated by another job: ../RNASeq/results/featureCounts/exp2.txt
wildcards: sampleName=exp2
resources: tmpdir=/tmp
[Wed Jul 13 18:19:50 2022]
rule RNASeq_mockDifferentialAnalysis:
input: ../RNASeq/results/featureCounts/exp1.txt
output: ../RNASeq/results/differentialAnalysis/exp1.csv
jobid: 3
reason: Missing output files: ../RNASeq/results/differentialAnalysis/exp1.csv; Input files updated by another job: ../RNASeq/results/featureCounts/exp1.txt
wildcards: sampleName=exp1
resources: tmpdir=/tmp
[Wed Jul 13 18:19:50 2022]
localrule RNASeq_all:
input: ../RNASeq/results/featureCounts/exp1.txt, ../RNASeq/results/featureCounts/exp2.txt, ../RNASeq/results/differentialAnalysis/exp1.csv, ../RNASeq/results/differentialAnalysis/exp2.csv
jobid: 0
reason: Input files updated by another job: ../RNASeq/results/differentialAnalysis/exp1.csv, ../RNASeq/results/differentialAnalysis/exp2.csv, ../RNASeq/results/featureCounts/exp2.txt, ../RNASeq/results/featureCounts/exp1.txt
resources: tmpdir=/tmp
Job stats:
job count min threads max threads
------------------------------- ------- ------------- -------------
RNASeq_all 1 1 1
RNASeq_mockDifferentialAnalysis 2 1 1
RNASeq_mockFeatureCounts 2 1 1
total 5 1 1
This was a dry-run (flag -n). The order of jobs does not reflect the order of execution.You see that only the RNAseq subworkflow was run. To collect both the targets of RNAseq and ChIPSeq, we need to add the instruction default_target: True to rule all. Modify Snakefile-MultiOmics as follows:
from snakemake.utils import min_version
min_version("6.0.0")
onstart:
print("##### ChIP-seq and RNA-seq workflows #####\n")
print("\t Reading samples and metadata....\n")
module RNASeq:
prefix: "../RNASeq"
snakefile:
"../RNASeq/Snakefile-RNASeq"
## This instruction should come right after the module
use rule * from RNASeq as RNASeq_*
module ChIPSeq:
prefix: "../ChIPSeq"
snakefile:
"../ChIPSeq/Snakefile-ChIPSeq"
## This instruction should come right after the module
use rule * from ChIPSeq as ChIPSeq_*
rule all:
input:
rules.RNASeq_all.input,
rules.ChIPSeq_all.input
default_target: TruePerform a real run:
#!/usr/bin/bash
snakemake --snakefile Snakefile-MultiOmics --cores 1
You should obtain:
Building DAG of jobs...
Using shell: /usr/bin/bash
Provided cores: 1 (use --cores to define parallelism)
Rules claiming more threads will be scaled down.
Job stats:
job count min threads max threads
------------------------------- ------- ------------- -------------
ChIPSeq_mockAlignment 2 1 1
ChIPSeq_mockpeakDetection 2 1 1
RNASeq_mockDifferentialAnalysis 2 1 1
RNASeq_mockFeatureCounts 2 1 1
all 1 1 1
total 9 1 1
##### ChIP-seq and RNA-seq workflows #####
Reading samples and metadata....
Select jobs to execute...
[Wed Jul 13 18:22:54 2022]
rule RNASeq_mockFeatureCounts:
input: ../RNASeq/inputs/exp1.fastq
output: ../RNASeq/results/featureCounts/exp1.txt
jobid: 1
reason: Missing output files: ../RNASeq/results/featureCounts/exp1.txt
wildcards: sampleName=exp1
resources: tmpdir=/tmp
[Wed Jul 13 18:23:04 2022]
Finished job 1.
1 of 9 steps (11%) done
Select jobs to execute...
[Wed Jul 13 18:23:04 2022]
rule RNASeq_mockDifferentialAnalysis:
input: ../RNASeq/results/featureCounts/exp1.txt
output: ../RNASeq/results/differentialAnalysis/exp1.csv
jobid: 3
reason: Missing output files: ../RNASeq/results/differentialAnalysis/exp1.csv; Input files updated by another job: ../RNASeq/results/featureCounts/exp1.txt
wildcards: sampleName=exp1
resources: tmpdir=/tmp
[Wed Jul 13 18:23:04 2022]
Error in rule RNASeq_mockDifferentialAnalysis:
jobid: 3
output: ../RNASeq/results/differentialAnalysis/exp1.csv
RuleException:
NameError in line 8 of /g/romebioinfo/tmp/demo-subworkflows/RNASeq/external_rules/callscript.smk:
name 'config' is not defined
File "/g/romebioinfo/tmp/demo-subworkflows/RNASeq/external_rules/callscript.smk", line 8, in __rule_mockDifferentialAnalysis
File "/g/romebioinfo/tmp/demo-subworkflows/envs/smake/lib/python3.9/concurrent/futures/thread.py", line 58, in run
Shutting down, this might take some time.
Exiting because a job execution failed. Look above for error message
Complete log: .snakemake/log/2022-07-13T182254.046552.snakemake.logNotice the error:
RuleException:
NameError in line 8 of /g/romebioinfo/tmp/demo-subworkflows/RNASeq/external_rules/callscript.smk:
name 'config' is not defined
File "/g/romebioinfo/tmp/demo-subworkflows/RNASeq/external_rules/callscript.smk", line 8, in __rule_mockDifferentialAnalysis
File "/g/romebioinfo/tmp/demo-subworkflows/envs/smake/lib/python3.9/concurrent/futures/thread.py", line 58, in runFor some reasons, when calling a script, snakemake is looking for a config file even if it is not used in the subworkflows per se. For this reason, create a config.yaml in the current folder with the following empty fields:
RNASeq:
summaryFile: ""
ChIPSeq:
summaryFile: ""Now add the config instruction to Snakefile-MultiOmics as follows:
from snakemake.utils import min_version
min_version("6.0.0")
onstart:
print("##### ChIP-seq and RNA-seq workflows #####\n")
print("\t Reading samples and metadata....\n")
module RNASeq:
prefix: "../RNASeq"
snakefile:
"../RNASeq/Snakefile-RNASeq"
config:
config["RNASeq"]
## This instruction should come right after the module
use rule * from RNASeq as RNASeq_*
module ChIPSeq:
prefix: "../ChIPSeq"
snakefile:
"../ChIPSeq/Snakefile-ChIPSeq"
config:
config["ChIPSeq"]
## This instruction should come right after the module
use rule * from ChIPSeq as ChIPSeq_*
rule all:
input:
rules.RNASeq_all.input,
rules.ChIPSeq_all.input
default_target: True
Perform a real run giving the configuration file as a parameter:
#!/usr/bin/bash
snakemake --snakefile Snakefile-MultiOmics --cores 1 --configfile config.yaml
You should get:
Building DAG of jobs...
Using shell: /usr/bin/bash
Provided cores: 1 (use --cores to define parallelism)
Rules claiming more threads will be scaled down.
Job stats:
job count min threads max threads
------------------------------- ------- ------------- -------------
ChIPSeq_mockAlignment 2 1 1
ChIPSeq_mockpeakDetection 2 1 1
RNASeq_mockDifferentialAnalysis 2 1 1
RNASeq_mockFeatureCounts 1 1 1
all 1 1 1
total 8 1 1
##### ChIP-seq and RNA-seq workflows #####
Reading samples and metadata....
Select jobs to execute...
[Wed Jul 13 20:11:17 2022]
rule RNASeq_mockDifferentialAnalysis:
input: ../RNASeq/results/featureCounts/exp1.txt
output: ../RNASeq/results/differentialAnalysis/exp1.csv
jobid: 3
reason: Missing output files: ../RNASeq/results/differentialAnalysis/exp1.csv
wildcards: sampleName=exp1
resources: tmpdir=/tmp
Writing to ../RNASeq/results/differentialAnalysis/exp1.csv
[Wed Jul 13 20:11:22 2022]
Finished job 3.
1 of 8 steps (12%) done
Select jobs to execute...
[Wed Jul 13 20:11:22 2022]
rule RNASeq_mockFeatureCounts:
input: ../RNASeq/inputs/exp2.fastq
output: ../RNASeq/results/featureCounts/exp2.txt
jobid: 2
reason: Missing output files: ../RNASeq/results/featureCounts/exp2.txt
wildcards: sampleName=exp2
resources: tmpdir=/tmp
[Wed Jul 13 20:11:32 2022]
Finished job 2.
2 of 8 steps (25%) done
Select jobs to execute...
[Wed Jul 13 20:11:33 2022]
rule ChIPSeq_mockAlignment:
input: ../ChIPSeq/inputs/exp2.fastq
output: ../ChIPSeq/results/alignment/exp2.bam
jobid: 6
reason: Missing output files: ../ChIPSeq/results/alignment/exp2.bam
wildcards: sampleName=exp2
resources: tmpdir=/tmp
[Wed Jul 13 20:11:43 2022]
Finished job 6.
3 of 8 steps (38%) done
Select jobs to execute...
[Wed Jul 13 20:11:43 2022]
rule ChIPSeq_mockpeakDetection:
input: ../ChIPSeq/results/alignment/exp2.bam
output: ../ChIPSeq/results/peaks/exp2.peaks
jobid: 8
reason: Missing output files: ../ChIPSeq/results/peaks/exp2.peaks; Input files updated by another job: ../ChIPSeq/results/alignment/exp2.bam
wildcards: sampleName=exp2
resources: tmpdir=/tmp
Writing to ../ChIPSeq/results/peaks/exp2.peaks
[Wed Jul 13 20:11:46 2022]
Finished job 8.
4 of 8 steps (50%) done
Select jobs to execute...
[Wed Jul 13 20:11:46 2022]
rule RNASeq_mockDifferentialAnalysis:
input: ../RNASeq/results/featureCounts/exp2.txt
output: ../RNASeq/results/differentialAnalysis/exp2.csv
jobid: 4
reason: Missing output files: ../RNASeq/results/differentialAnalysis/exp2.csv; Input files updated by another job: ../RNASeq/results/featureCounts/exp2.txt
wildcards: sampleName=exp2
resources: tmpdir=/tmp
Writing to ../RNASeq/results/differentialAnalysis/exp2.csv
[Wed Jul 13 20:11:48 2022]
Finished job 4.
5 of 8 steps (62%) done
Select jobs to execute...
[Wed Jul 13 20:11:48 2022]
rule ChIPSeq_mockAlignment:
input: ../ChIPSeq/inputs/exp1.fastq
output: ../ChIPSeq/results/alignment/exp1.bam
jobid: 5
reason: Missing output files: ../ChIPSeq/results/alignment/exp1.bam
wildcards: sampleName=exp1
resources: tmpdir=/tmp
[Wed Jul 13 20:11:59 2022]
Finished job 5.
6 of 8 steps (75%) done
Select jobs to execute...
[Wed Jul 13 20:11:59 2022]
rule ChIPSeq_mockpeakDetection:
input: ../ChIPSeq/results/alignment/exp1.bam
output: ../ChIPSeq/results/peaks/exp1.peaks
jobid: 7
reason: Missing output files: ../ChIPSeq/results/peaks/exp1.peaks; Input files updated by another job: ../ChIPSeq/results/alignment/exp1.bam
wildcards: sampleName=exp1
resources: tmpdir=/tmp
Writing to ../ChIPSeq/results/peaks/exp1.peaks
[Wed Jul 13 20:12:01 2022]
Finished job 7.
7 of 8 steps (88%) done
Select jobs to execute...
[Wed Jul 13 20:12:01 2022]
localrule all:
input: ../RNASeq/results/featureCounts/exp1.txt, ../RNASeq/results/featureCounts/exp2.txt, ../RNASeq/results/differentialAnalysis/exp1.csv, ../RNASeq/results/differentialAnalysis/exp2.csv, ../ChIPSeq/results/alignment/exp1.bam, ../ChIPSeq/results/alignment/exp2.bam, ../ChIPSeq/results/peaks/exp1.peaks, ../ChIPSeq/results/peaks/exp2.peaks
jobid: 0
reason: Input files updated by another job: ../ChIPSeq/results/alignment/exp1.bam, ../RNASeq/results/differentialAnalysis/exp2.csv, ../ChIPSeq/results/alignment/exp2.bam, ../ChIPSeq/results/peaks/exp1.peaks, ../RNASeq/results/differentialAnalysis/exp1.csv, ../ChIPSeq/results/peaks/exp2.peaks, ../RNASeq/results/featureCounts/exp2.txt
resources: tmpdir=/tmp
[Wed Jul 13 20:12:01 2022]
Finished job 0.
8 of 8 steps (100%) done
Complete log: .snakemake/log/2022-07-13T201116.342815.snakemake.log
Running MultiOmics on a HPC
Let’s start by deleting the previously created results to be able to run the workflow again:
#!/usr/bin/bash
rm -r ../RNASeq/results
rm -r ../ChIPSeq/results
As we did for the other workflows, copy the following content to profileMultiOmics/config.yaml:
---
snakefile: Snakefile-MultiOmics
latency-wait: 60
reason: True
show-failed-logs: True
keep-going: True
printshellcmds: True
configfile: "config.yaml"
# Cluster submission
jobname: "{rule}.{jobid}" # Provide a custom name for the jobscript that is submitted to the cluster.
max-jobs-per-second: 1 #Maximal number of cluster/drmaa jobs per second, default is 10, fractions allowed.
max-status-checks-per-second: 10 #Maximal number of job status checks per second, default is 10
jobs: 400 #Use at most N CPU cluster/cloud jobs in parallel.
cluster: "sbatch --output=\"jobs/{rule}/slurm_%x_%j.out\" --error=\"jobs/{rule}/slurm_%x_%j.log\" --mem={resources.mem_mb} --time={resources.runtime} --parsable"
cluster-status: "./profileMultiOmics/status-sacct.sh" # Use to handle timeout exception, do not forget to chmod +x
Exactly as before, add profileMultiOmics/status-sacct.sh:
#!/usr/bin/env bash
# Check status of Slurm job
jobid="$1"
if [[ "$jobid" == Submitted ]]
then
echo smk-simple-slurm: Invalid job ID: "$jobid" >&2
echo smk-simple-slurm: Did you remember to add the flag --parsable to your sbatch call? >&2
exit 1
fi
output=`sacct -j "$jobid" --format State --noheader | head -n 1 | awk '{print $1}'`
if [[ $output =~ ^(COMPLETED).* ]]
then
echo success
elif [[ $output =~ ^(RUNNING|PENDING|COMPLETING|CONFIGURING|SUSPENDED).* ]]
then
echo running
else
echo failed
fi
Make it executable:
#!/usr/bin/bash
chmod +x profileMultiOmics/status-sacct.sh
Perform a real run:
#!/usr/bin/bash
snakemake --profile profileMultiOmics/
You should get the error:
WorkflowError:
Error when formatting 'sbatch --output="jobs/{rule}/slurm_%x_%j.out" --error="jobs/{rule}/slurm_%x_%j.log" --mem={resources.mem_mb} --time={resources.runtime} --parsable' for rule ChIPSeq_mockAlignment. 'Resources' object has no attribute 'runtime'
It cannot find the runtime required for the rule ChIPSeq_mockAlignment. Indeed we did not define the resources used for each job as we did in the previous parts. Attention: The prefixes must be added to the job’s names. Copy the resources definition of each subworkflow to profileMultiOmics/config.yaml. Take care of adding the corresponding prefixes:
---
snakefile: Snakefile-MultiOmics
latency-wait: 60
reason: True
show-failed-logs: True
keep-going: True
printshellcmds: True
configfile: "config.yaml"
# Cluster submission
jobname: "{rule}.{jobid}" # Provide a custom name for the jobscript that is submitted to the cluster.
max-jobs-per-second: 1 #Maximal number of cluster/drmaa jobs per second, default is 10, fractions allowed.
max-status-checks-per-second: 10 #Maximal number of job status checks per second, default is 10
jobs: 400 #Use at most N CPU cluster/cloud jobs in parallel.
cluster: "sbatch --output=\"jobs/{rule}/slurm_%x_%j.out\" --error=\"jobs/{rule}/slurm_%x_%j.log\" --mem={resources.mem_mb} --time={resources.runtime} --parsable"
cluster-status: "./profileMultiOmics/status-sacct.sh" # Use to handle timeout exception, do not forget to chmod +x
# Job resources
set-resources:
- RNASeq_mockFeatureCounts:mem_mb=1000
- RNASeq_mockFeatureCounts:runtime=00:03:00
- RNASeq_mockDifferentialAnalysis:mem_mb=1000
- RNASeq_mockDifferentialAnalysis:runtime=00:03:00
- ChIPSeq_mockAlignment:mem_mb=1000
- ChIPSeq_mockAlignment:runtime=00:03:00
- ChIPSeq_mockpeakDetection:mem_mb=1000
- ChIPSeq_mockpeakDetection:runtime=00:03:00
# For some reasons time needs quotes to be read by snakemake
default-resources:
- mem_mb=500
- runtime="00:01:00"
# Define the number of threads used by rules
set-threads:
- RNASeq_mockFeatureCounts=1
- RNASeq_mockDifferentialAnalysis=1
- ChIPSeq_mockAlignment=1
- ChIPSeq_mockpeakDetection=1
Perform a real run:
#!/usr/bin/bash
snakemake --profile profileMultiOmics/
You should get:
Building DAG of jobs...
Using shell: /usr/bin/bash
Provided cluster nodes: 400
Job stats:
job count min threads max threads
------------------------------- ------- ------------- -------------
ChIPSeq_mockAlignment 2 1 1
ChIPSeq_mockpeakDetection 2 1 1
RNASeq_mockDifferentialAnalysis 2 1 1
RNASeq_mockFeatureCounts 2 1 1
all 1 1 1
total 9 1 1
##### ChIP-seq and RNA-seq workflows #####
Reading samples and metadata....
Select jobs to execute...
[Wed Jul 13 20:45:22 2022]
rule ChIPSeq_mockAlignment:
input: ../ChIPSeq/inputs/exp1.fastq
output: ../ChIPSeq/results/alignment/exp1.bam
jobid: 5
reason: Missing output files: ../ChIPSeq/results/alignment/exp1.bam
wildcards: sampleName=exp1
resources: mem_mb=1000, disk_mb=1000, tmpdir=/tmp, runtime=00:03:00
cat ../ChIPSeq/inputs/exp1.fastq > ../ChIPSeq/results/alignment/exp1.bam
sleep 10s
Submitted job 5 with external jobid '44401149'.
[Wed Jul 13 20:45:22 2022]
rule ChIPSeq_mockAlignment:
input: ../ChIPSeq/inputs/exp2.fastq
output: ../ChIPSeq/results/alignment/exp2.bam
jobid: 6
reason: Missing output files: ../ChIPSeq/results/alignment/exp2.bam
wildcards: sampleName=exp2
resources: mem_mb=1000, disk_mb=1000, tmpdir=/tmp, runtime=00:03:00
cat ../ChIPSeq/inputs/exp2.fastq > ../ChIPSeq/results/alignment/exp2.bam
sleep 10s
Submitted job 6 with external jobid '44401150'.
[Wed Jul 13 20:45:22 2022]
rule RNASeq_mockFeatureCounts:
input: ../RNASeq/inputs/exp2.fastq
output: ../RNASeq/results/featureCounts/exp2.txt
jobid: 2
reason: Missing output files: ../RNASeq/results/featureCounts/exp2.txt
wildcards: sampleName=exp2
resources: mem_mb=1000, disk_mb=1000, tmpdir=/tmp, runtime=00:03:00
cat ../RNASeq/inputs/exp2.fastq > ../RNASeq/results/featureCounts/exp2.txt
sleep 10s
Submitted job 2 with external jobid '44401151'.
[Wed Jul 13 20:45:22 2022]
rule RNASeq_mockFeatureCounts:
input: ../RNASeq/inputs/exp1.fastq
output: ../RNASeq/results/featureCounts/exp1.txt
jobid: 1
reason: Missing output files: ../RNASeq/results/featureCounts/exp1.txt
wildcards: sampleName=exp1
resources: mem_mb=1000, disk_mb=1000, tmpdir=/tmp, runtime=00:03:00
cat ../RNASeq/inputs/exp1.fastq > ../RNASeq/results/featureCounts/exp1.txt
sleep 10s
Submitted job 1 with external jobid '44401152'.
[Wed Jul 13 20:46:24 2022]
Error in rule ChIPSeq_mockAlignment:
jobid: 5
output: ../ChIPSeq/results/alignment/exp1.bam
shell:
cat ../ChIPSeq/inputs/exp1.fastq > ../ChIPSeq/results/alignment/exp1.bam
sleep 10s
(one of the commands exited with non-zero exit code; note that snakemake uses bash strict mode!)
cluster_jobid: 44401149
Error executing rule ChIPSeq_mockAlignment on cluster (jobid: 5, external: 44401149, jobscript: /g/romebioinfo/tmp/demo-subworkflows/MultiOmics/.snakemake/tmp.lvuuylhl/ChIPSeq_mockAlignment.5). For error details see the cluster log and the log files of the involved rule(s).
[Wed Jul 13 20:46:24 2022]
Error in rule ChIPSeq_mockAlignment:
jobid: 6
output: ../ChIPSeq/results/alignment/exp2.bam
shell:
cat ../ChIPSeq/inputs/exp2.fastq > ../ChIPSeq/results/alignment/exp2.bam
sleep 10s
(one of the commands exited with non-zero exit code; note that snakemake uses bash strict mode!)
cluster_jobid: 44401150
Error executing rule ChIPSeq_mockAlignment on cluster (jobid: 6, external: 44401150, jobscript: /g/romebioinfo/tmp/demo-subworkflows/MultiOmics/.snakemake/tmp.lvuuylhl/ChIPSeq_mockAlignment.6). For error details see the cluster log and the log files of the involved rule(s).
[Wed Jul 13 20:46:24 2022]
Error in rule RNASeq_mockFeatureCounts:
jobid: 2
output: ../RNASeq/results/featureCounts/exp2.txt
shell:
cat ../RNASeq/inputs/exp2.fastq > ../RNASeq/results/featureCounts/exp2.txt
sleep 10s
(one of the commands exited with non-zero exit code; note that snakemake uses bash strict mode!)
cluster_jobid: 44401151
Error executing rule RNASeq_mockFeatureCounts on cluster (jobid: 2, external: 44401151, jobscript: /g/romebioinfo/tmp/demo-subworkflows/MultiOmics/.snakemake/tmp.lvuuylhl/RNASeq_mockFeatureCounts.2). For error details see the cluster log and the log files of the involved rule(s).
[Wed Jul 13 20:46:24 2022]
Error in rule RNASeq_mockFeatureCounts:
jobid: 1
output: ../RNASeq/results/featureCounts/exp1.txt
shell:
cat ../RNASeq/inputs/exp1.fastq > ../RNASeq/results/featureCounts/exp1.txt
sleep 10s
(one of the commands exited with non-zero exit code; note that snakemake uses bash strict mode!)
cluster_jobid: 44401152
Error executing rule RNASeq_mockFeatureCounts on cluster (jobid: 1, external: 44401152, jobscript: /g/romebioinfo/tmp/demo-subworkflows/MultiOmics/.snakemake/tmp.lvuuylhl/RNASeq_mockFeatureCounts.1). For error details see the cluster log and the log files of the involved rule(s).
Exiting because a job execution failed. Look above for error message
Complete log: .snakemake/log/2022-07-13T204521.378559.snakemake.log
You can see several errors thrown by snakemake. It indicates checking the log files or the cluster logs. However, you will notice that the jobs folder was not created. This is causing snakemake to not be able to write log files and causes it to fail. Delete the on start: sections of ../RNASeq/snakemake-RNASeq and ../ChIPSeq/snakemake-ChIPSeq. Add the creation of the relevant folders to the on start: section in Snakefile-MultiOmics as follows. Note that you must add the prefixes to the output folders.
from snakemake.utils import min_version
min_version("6.0.0")
onstart:
print("##### ChIP-seq and RNA-seq workflows #####\n")
print("\t Reading samples and metadata....\n")
shell("mkdir -p jobs/ChIPSeq_mockAlignment")
shell("mkdir -p jobs/ChIPSeq_mockpeakDetection")
shell("mkdir -p jobs/RNASeq_mockFeatureCounts")
shell("mkdir -p jobs/RNASeq_mockDifferentialAnalysis")
module RNASeq:
prefix: "../RNASeq"
snakefile:
"../RNASeq/Snakefile-RNASeq"
config:
config["RNASeq"]
## This instruction should come right after the module
use rule * from RNASeq as RNASeq_*
module ChIPSeq:
prefix: "../ChIPSeq"
snakefile:
"../ChIPSeq/Snakefile-ChIPSeq"
config:
config["ChIPSeq"]
## This instruction should come right after the module
use rule * from ChIPSeq as ChIPSeq_*
rule all:
input:
rules.RNASeq_all.input,
rules.ChIPSeq_all.input
default_target: True
Perform a real run:
#!/usr/bin/bash
snakemake --profile profileMultiOmics/
The pipeline should now terminate without errors. You should get:
Building DAG of jobs...
Using shell: /usr/bin/bash
Provided cluster nodes: 400
Job stats:
job count min threads max threads
------------------------------- ------- ------------- -------------
ChIPSeq_mockAlignment 2 1 1
ChIPSeq_mockpeakDetection 2 1 1
RNASeq_mockDifferentialAnalysis 2 1 1
RNASeq_mockFeatureCounts 2 1 1
all 1 1 1
total 9 1 1
##### ChIP-seq and RNA-seq workflows #####
Reading samples and metadata....
mkdir -p jobs/ChIPSeq_mockAlignment
mkdir -p jobs/ChIPSeq_mockpeakDetection
mkdir -p jobs/RNASeq_mockFeatureCounts
mkdir -p jobs/RNASeq_mockDifferentialAnalysis
Select jobs to execute...
[Wed Jul 13 21:02:50 2022]
rule ChIPSeq_mockAlignment:
input: ../ChIPSeq/inputs/exp1.fastq
output: ../ChIPSeq/results/alignment/exp1.bam
jobid: 5
reason: Missing output files: ../ChIPSeq/results/alignment/exp1.bam
wildcards: sampleName=exp1
resources: mem_mb=1000, disk_mb=1000, tmpdir=/tmp, runtime=00:03:00
cat ../ChIPSeq/inputs/exp1.fastq > ../ChIPSeq/results/alignment/exp1.bam
sleep 10s
Submitted job 5 with external jobid '44401764'.
[Wed Jul 13 21:02:51 2022]
rule ChIPSeq_mockAlignment:
input: ../ChIPSeq/inputs/exp2.fastq
output: ../ChIPSeq/results/alignment/exp2.bam
jobid: 6
reason: Missing output files: ../ChIPSeq/results/alignment/exp2.bam
wildcards: sampleName=exp2
resources: mem_mb=1000, disk_mb=1000, tmpdir=/tmp, runtime=00:03:00
cat ../ChIPSeq/inputs/exp2.fastq > ../ChIPSeq/results/alignment/exp2.bam
sleep 10s
Submitted job 6 with external jobid '44401766'.
[Wed Jul 13 21:02:51 2022]
rule RNASeq_mockFeatureCounts:
input: ../RNASeq/inputs/exp2.fastq
output: ../RNASeq/results/featureCounts/exp2.txt
jobid: 2
reason: Missing output files: ../RNASeq/results/featureCounts/exp2.txt
wildcards: sampleName=exp2
resources: mem_mb=1000, disk_mb=1000, tmpdir=/tmp, runtime=00:03:00
cat ../RNASeq/inputs/exp2.fastq > ../RNASeq/results/featureCounts/exp2.txt
sleep 10s
Submitted job 2 with external jobid '44401767'.
[Wed Jul 13 21:02:51 2022]
rule RNASeq_mockFeatureCounts:
input: ../RNASeq/inputs/exp1.fastq
output: ../RNASeq/results/featureCounts/exp1.txt
jobid: 1
reason: Missing output files: ../RNASeq/results/featureCounts/exp1.txt
wildcards: sampleName=exp1
resources: mem_mb=1000, disk_mb=1000, tmpdir=/tmp, runtime=00:03:00
cat ../RNASeq/inputs/exp1.fastq > ../RNASeq/results/featureCounts/exp1.txt
sleep 10s
Submitted job 1 with external jobid '44401768'.
[Wed Jul 13 21:03:41 2022]
Finished job 5.
1 of 9 steps (11%) done
Select jobs to execute...
[Wed Jul 13 21:03:41 2022]
rule ChIPSeq_mockpeakDetection:
input: ../ChIPSeq/results/alignment/exp1.bam
output: ../ChIPSeq/results/peaks/exp1.peaks
jobid: 7
reason: Missing output files: ../ChIPSeq/results/peaks/exp1.peaks; Input files updated by another job: ../ChIPSeq/results/alignment/exp1.bam
wildcards: sampleName=exp1
resources: mem_mb=1000, disk_mb=1000, tmpdir=/tmp, runtime=00:03:00
Submitted job 7 with external jobid '44401828'.
[Wed Jul 13 21:03:42 2022]
Finished job 6.
2 of 9 steps (22%) done
Waiting at most 60 seconds for missing files.
[Wed Jul 13 21:03:51 2022]
Finished job 2.
3 of 9 steps (33%) done
[Wed Jul 13 21:03:51 2022]
Finished job 1.
4 of 9 steps (44%) done
Select jobs to execute...
[Wed Jul 13 21:03:52 2022]
rule RNASeq_mockDifferentialAnalysis:
input: ../RNASeq/results/featureCounts/exp2.txt
output: ../RNASeq/results/differentialAnalysis/exp2.csv
jobid: 4
reason: Missing output files: ../RNASeq/results/differentialAnalysis/exp2.csv; Input files updated by another job: ../RNASeq/results/featureCounts/exp2.txt
wildcards: sampleName=exp2
resources: mem_mb=1000, disk_mb=1000, tmpdir=/tmp, runtime=00:03:00
Submitted job 4 with external jobid '44401835'.
[Wed Jul 13 21:03:53 2022]
rule ChIPSeq_mockpeakDetection:
input: ../ChIPSeq/results/alignment/exp2.bam
output: ../ChIPSeq/results/peaks/exp2.peaks
jobid: 8
reason: Missing output files: ../ChIPSeq/results/peaks/exp2.peaks; Input files updated by another job: ../ChIPSeq/results/alignment/exp2.bam
wildcards: sampleName=exp2
resources: mem_mb=1000, disk_mb=1000, tmpdir=/tmp, runtime=00:03:00
Submitted job 8 with external jobid '44401837'.
[Wed Jul 13 21:03:53 2022]
rule RNASeq_mockDifferentialAnalysis:
input: ../RNASeq/results/featureCounts/exp1.txt
output: ../RNASeq/results/differentialAnalysis/exp1.csv
jobid: 3
reason: Missing output files: ../RNASeq/results/differentialAnalysis/exp1.csv; Input files updated by another job: ../RNASeq/results/featureCounts/exp1.txt
wildcards: sampleName=exp1
resources: mem_mb=1000, disk_mb=1000, tmpdir=/tmp, runtime=00:03:00
Submitted job 3 with external jobid '44401838'.
[Wed Jul 13 21:04:34 2022]
Finished job 3.
5 of 9 steps (56%) done
[Wed Jul 13 21:04:44 2022]
Finished job 7.
6 of 9 steps (67%) done
[Wed Jul 13 21:04:44 2022]
Finished job 4.
7 of 9 steps (78%) done
[Wed Jul 13 21:04:44 2022]
Finished job 8.
8 of 9 steps (89%) done
Select jobs to execute...
[Wed Jul 13 21:04:44 2022]
localrule all:
input: ../RNASeq/results/featureCounts/exp1.txt, ../RNASeq/results/featureCounts/exp2.txt, ../RNASeq/results/differentialAnalysis/exp1.csv, ../RNASeq/results/differentialAnalysis/exp2.csv, ../ChIPSeq/results/alignment/exp1.bam, ../ChIPSeq/results/alignment/exp2.bam, ../ChIPSeq/results/peaks/exp1.peaks, ../ChIPSeq/results/peaks/exp2.peaks
jobid: 0
reason: Input files updated by another job: ../ChIPSeq/results/alignment/exp2.bam, ../RNASeq/results/differentialAnalysis/exp1.csv, ../RNASeq/results/featureCounts/exp2.txt, ../ChIPSeq/results/alignment/exp1.bam, ../ChIPSeq/results/peaks/exp1.peaks, ../RNASeq/results/differentialAnalysis/exp2.csv, ../RNASeq/results/featureCounts/exp1.txt, ../ChIPSeq/results/peaks/exp2.peaks
resources: mem_mb=500, disk_mb=1000, tmpdir=/tmp, runtime=00:01:00
[Wed Jul 13 21:04:44 2022]
Finished job 0.
9 of 9 steps (100%) done
Complete log: .snakemake/log/2022-07-13T210249.161330.snakemake.log
Acknowledgements
Thank you to Thomas Weber for his comments and suggestions.