Deutsch
Deutsch Čeština
Čeština Ελληνικά
Ελληνικά
TreeTOPS – a phylogeny icebreaker game
Introduction
Playful introduction to phylogentic trees
This icebreaker game aims to introduce the players to phylogenetics – the study of evolutionary relationships among taxonomic groups (e.g., organisms, species and populations) or other biological units (e.g., genes and proteins). Using short pieces of colour patterns, the players work in a group and build their own phylogenetic tree to illustrate the evolutionary branching process of an imaginary taxonomic group or biological unit.
Background
Classical versus molecular phylogenetics
Phylogenetic reconstruction has been playing a key role in understanding evolutionary processes. Traditional classification methods have looked at the evolutionary relationships of classical biological units like phyla, species or populations, and were based on morphological observations and phenotypic characteristics. Fast-paced advances in molecular biology within the past decades, however, have revolutionised the phylogenetic study of organisms as well as of their molecules. Techniques like the polymerase chain reaction (PCR), restriction fragment length polymorphism (RFLP) and, most recently, DNA and whole genome sequencing, coupled with an increase in biological databases, have significantly extended the scope of phylogenetics and allowed scientists to build evolutionary trees on the basis of the molecular make up of organisms. Through these developments, molecular data has now become the prime source for the construction of phylogenetic trees.
There are numerous examples of how the use of molecular data for phylogenetics has advanced the study of evolution. For instance, until the 1970s, fungi were classified as a subkingdom of plants. It was only through analysis of molecular data that it emerged that fungi are actually more closely related to animals than plants. This led to a dramatic reclassification of fungi into their own distinct kingdom. Molecular sequence data also allowed scientists to establish classification systems for viruses, genes and proteins, providing insights relevant to the whole spectrum of biological and medical sciences from evolution to proteomics and medicine. With the use of molecular data for the assessment of relatedness, identification of taxonomic common ancestors has become more accurate. This advancement not only offers vital clues on the evolutionary relationship of organisms but can also help identify new model organism or organisms which are the closest resembling relatives to ancient common ancestors.
Tree basics
In phylogenetic trees, the degree of evolutionary relatedness is expressed in the positions of the individual units of interest (e.g., species, populations, genes) to each other. Units which are closely related are placed close together, while units which are more distantly related are positioned on different branches or clades in the tree. The text below compares two of the most common types of phylogenetic trees and defines some important terminologies.
Unrooted versus rooted phylogenetic trees
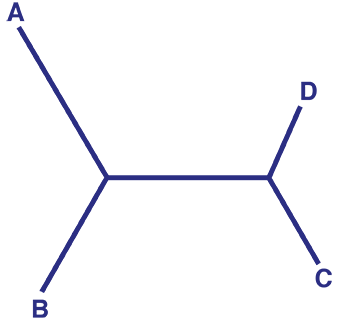
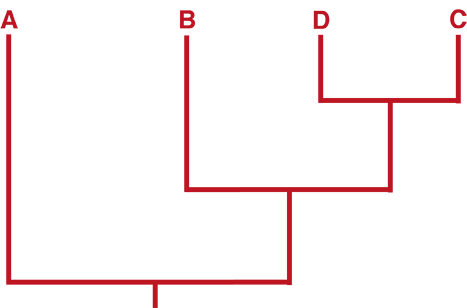
An unrooted tree does not provide any information about the common ancestor of the study group. An unrooted tree can be converted into a rooted tree – a tree which provides information about the common ancestor (root) of the study group. By comparing the trait of interest present in the study group (ingroup) with the trait present in a group which is only distantly related to the ingroup (outgroup), it can be determined whether the trait of interest is ancestral (present in common ancestor) or derived (not present in common ancestor).
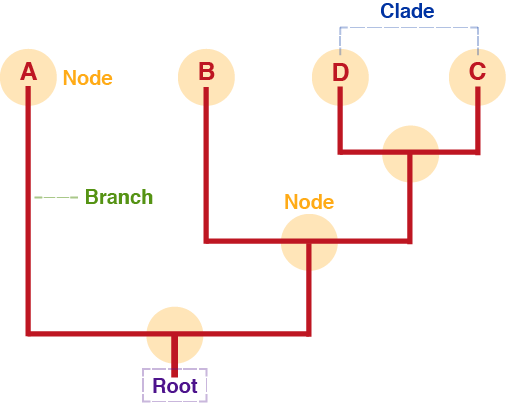
Phylogenetic terminology of rooted trees
Branch
Connects the nodes of the tree and represents the proposed evolutionary path of the specific lineage.
Root
Node which represents the common ancestor of all nodes in the tree.
Node
Internal node: represents hypothetical ancestor Terminal node (leaf): represents (usually present-day) taxon (population, species, gene, protein, etc.) which is being compared to other taxa in the tree.
Clade
In a clade, all members share a common ancestor which is not the common ancestor of any other member of the tree.
Challenges of building a tree
When assessing how biological units are related to each other, hypothetical evolutionary processes such as parallel, convergent or reticulate evolution cause the most common challenges. The possible occurrences of such evolutionary events within the evolution of the studied group must therefore be taken into consideration when building a phylogenetic tree. The following text briefly defines the characteristics of these three kinds of evolutionary processes.
Parallel evolution
Parallel evolution describes the process by which organisms, taxa, or biological units (e.g., genes or proteins) that are derived from a common ancestor have independently acquired comparable (analogous) traits. Parallel evolution can be the result of adapting to, e.g., the same ecological niche, or can be the result of other selective pressures.
Convergent evolution
Convergent evolution describes the process by which organisms, taxa, or other biological units (e.g., genes or proteins) that are not derived from a common ancestor have independently acquired comparable (analogous) traits. Convergent evolution can be the result of adapting to, e.g., the same ecological niche, or can be the result of other selective pressures.
Reticulate evolution
Reticulate evolution describes the process by which genetically distinct lineages/species recombine and a new species, which is reproductively isolated from the parent species, arises. This speciation event is called hybrid speciation. Considering reticulate evolution events when constructing a phylogenetic tree might mean that a strictly hierarchical tree structure does not appropriately reflect evolutionary relationships of this particular study group.
Similar to analysing the relatedness of classical biological units such as species or populations, studies of gene and protein evolution compare homologous DNA and amino acid sequences and functions to determine their evolutionary relationship. Homologous sequences share an identical or very similar sequence and have a common origin. The fact that there are several different ways in which homologues might arise provides a challenge when assessing relatedness. Again, it is therefore important to be aware of the possible evolutionary processes which might have played a role in the specific group of interest. The two most common types of homologues are defined below.
Orthologue
Homologous biological units (e.g., genes or proteins) are orthologues if they are found in different species and originate from the same ancestral biological unit (common ancestor). Orthologues arise through speciation from a common ancestor and have usually retained the same or similar function over the course of evolution.
Paralogue
Homologous biological units (e.g., genes or proteins) are paralogues if they are found in the same species (often the same organism) and have arisen through (recent or ancient) gene duplication within this species/organism. Over time, paralogues usually become different to each other in function (and sometimes sequence).
Game materials
Download the game material as pdf files by clicking here:
Game instructions
Material
- 8 (7) “descendant cards” (on printable magnets or laminated paper)
- 1 “common ancestor card” (on printable magnets or laminated paper)
- Magnetic white/black board or pin board covered by paper
- White board pen/chalk or paper pen
- Additional instructor’s material:
- Introduction and Background Sheet
- Solution Sheet
Players
8-9 (7-8) players depending on which game (game 1-3) is played.
Aim of the game
In this game, each playing card represents an imaginable biological unit. The aim of the game is to sort the cards according to their “relatedness” and construct a rooted phylogenetic tree using all the “descendant cards”, starting with the “common ancestor” card as root.
Note that there might be several possibilities how your phylogenetic tree could look like. The aim of the game will be fulfilled as long as decisions on the position of individual “descendant” within the tree can be justified when the players explain the tree structure to a parallel group or their instructor.
Hint
When deciding on the “hierarchies” and “groupings” within your tree, remember that on the genetic level evolution happens through small changes such as deletions, duplications, inversions and substitutions of nucleotides or nucleotide stretches.
Set up
8 (7) players: each participant randomly receives one of the “descendant cards”. The “common ancestor” card is placed at the bottom of the board as starting point of the phylogenetic tree.
9 (8) players: one participant receives the “common ancestor card”. All of the other participants randomly receive a “descendant card”.
Playing
1) Players group themselves according to their “descendant cards”. The player with the “common ancestor card” can assist in grouping the other players.
2) The “common ancestor card” is placed on the board. Once grouped, the players place their “descendant cards” on the board according to the structure the group has decided on.
3) Players draw a phylogram by connecting the individual cards.
4) Once the phylogenetic tree has been finalised, players explain their decisions with respect to the tree structure to participants of a TreeTOPS Game running in parallel or to their instructor.
Gallery




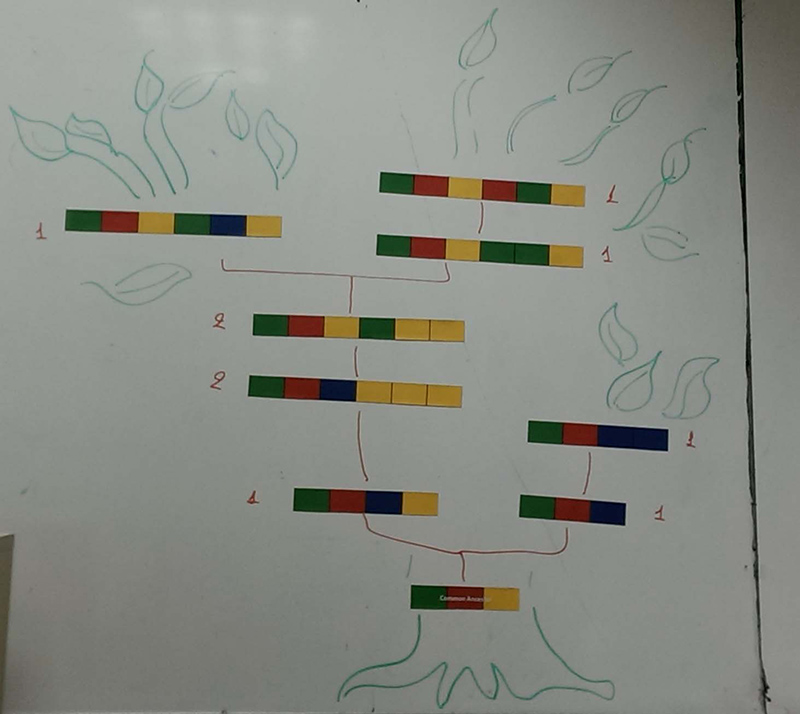
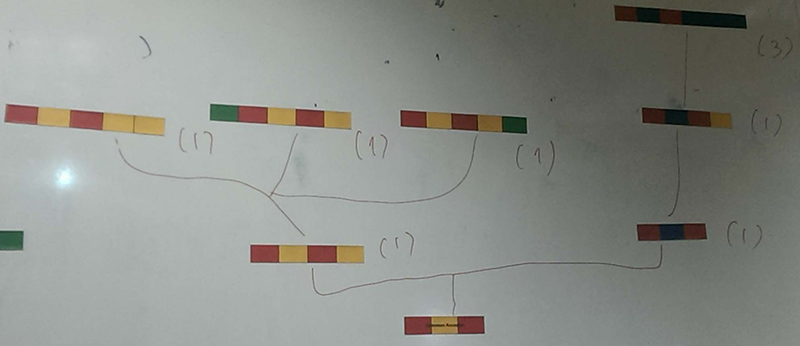

Topic area: Evolutionary biology, Genome biology
Age group: 16-19
Author: ELLS Team
Share: