Clement Blanchet
Team Leader
ORCID: 0000-0003-3432-8496
EditCharacterisation of biological molecules and assemblies using small angle x-ray scattering (SAXS)
Team Leader
ORCID: 0000-0003-3432-8496
EditBiological small angle X-ray scattering (BioSAXS) is a powerful technique to study the three-dimensional structures of biological macromolecules in solution. BioSAXS provides valuable insights into the size, shape, and overall architecture of proteins, nucleic acids, and lipid assemblies, including lipid nanoparticles. It seamlessly integrates with complementary methods such as MX, Cryo-EM, and NMR, enabling comprehensive investigations into protein folding, protein-protein interactions, intrinsically disordered proteins, and conformational changes. With its capability to analyse samples in their natural state, BioSAXS plays an important role in unraveling the intricate structure-function relationships of biological macromolecules and biomedical samples.
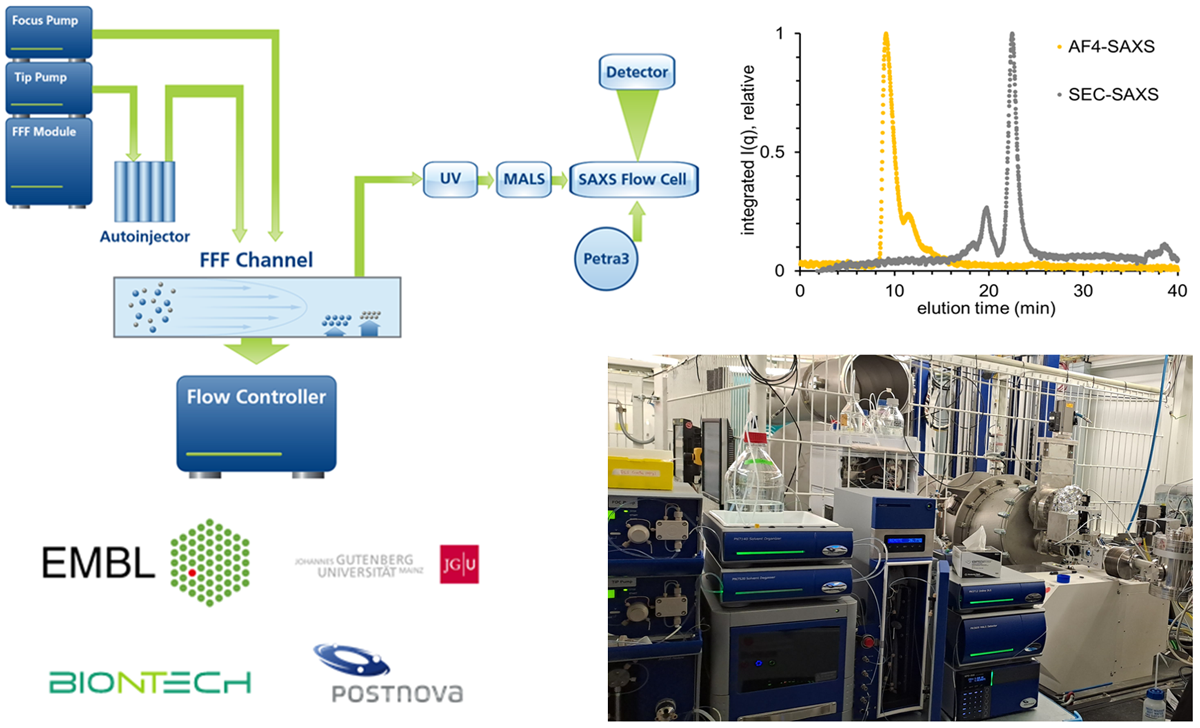
Our team operates and develops the P12 beamline, a cutting-edge instrument dedicated to biological small angle X-ray scattering (BioSAXS). The instrument routinely offers fully automated BioSAXS measurements of macromolecular solution, from sample handling to data analysis. The instrument routinely offers fully automated BioSAXS measurements of macromolecular solution, from sample handling to data analysis. Advanced scattering experiments, including anomalous- or time-resolved scattering, can also be carried out, leveraging the excellent beam properties of PETRAIII and the tunability of the beamline.
The instrument undergoes continuous development to enhance existing experiments and expand the range of capabilities offered by the beamline, enabling new and exciting research opportunities.
The team provides comprehensive support to academic and industrial users for data collection on the P12 beamline. In-depth collaborations for the analysis and interpretation of BioSAXS data are established when deemed relevant. Additionally, the team curates the SASBDB, a dedicated repository for SAXS data and models.
SAXS is ideal for 4D structural biology, as it involves studying biomolecules in solution, allowing easy modification. Through time-resolved investigations and high-throughput experiments, we can monitor induced structural changes, gaining comprehensive insights into the dynamic behavior and structural alterations of biomolecules.
To capture the dynamics of biomolecules and assemblies in solution, we will further develop time-resolved SAXS (TR-SAXS) techniques. By rapidly triggering specific perturbations, we can monitor the structural transitions and kinetics of biomolecular processes with high temporal resolution. This requires dedicated experimental setups, including fast mixing devices or light-triggered methods such as T-jump, caged compounds, and photo-activation. Through TR-SAXS, we aim to uncover essential insights into folding pathways, conformational changes, and functional mechanisms of biomolecules in real-time.
Our focus also extends to high-throughput SAXS (HT-SAXS) techniques, enabling comprehensive exploration of biomolecular structures under diverse conditions. We will develop dedicated sample environments, employing microfluidic technologies and automation to reduce sample requirements and measurement times. HT-SAXS facilitates rapid screening of various conditions, including pH, temperature, and ligand binding, unlocking multidimensional structural analysis possibilities. Our goal is to establish robust HT-SAXS methodologies and advanced data analysis pipelines for efficient exploration of the structural and dynamic properties of biomolecules in solution.
These advancements in SAXS for 4D structural biology will be accompanied by developments in SAXS data analysis methods. With the remarkable progress in computational structural biology, such as AlphaFold, a more comprehensive and generalised approach to SAXS data analysis based on predicted models can be established. These predicted models help evaluate the quality of experimental data and assess important sample parameters like size, shape, and flexibility. They can be validated or invalidated using SAXS data and enable the modelling of structures in oligomeric or complex assemblies, characterization of their flexibility, and exploration of alternative structural conformations. By combining computational predictions with experimental data, we can gain a deeper and more accurate understanding of biomolecular structures and dynamics.